The Lungs in Different Physiological States |
The increases in muscular oxygen consumption (![]() ) and carbon dioxide production (
) and carbon dioxide production (![]() ) accompanying whole-body exercise present a greater challenge to the maintenance of pulmonary gas exchange than any other physiologic stressor. This chapter discusses the responses of the healthy respiratory system to exercise with an emphasis on the following problems: What neurochemical mechanisms regulate the ventilatory response to exercise and what are the consequences of this hyperpnea to the work and to the fatigue of the respiratory muscles? What mechanisms underlie the widening of the alveolar to arterial partial pressure of oxygen (PO2) difference during exercise? How do the unique characteristics of the pulmonary circulation determine its response to exercise? How does respiration impact the cardiovascular response to exercise? Under what circumstances might the respiratory system provide a limitation to O2 transport and/or exercise performance? We consider these problems primarily in the healthy, young, normally fit adult, with reference to special cases of the highly trained athlete and to the effects of healthy aging, high altitude hypoxia, and physical training.
) accompanying whole-body exercise present a greater challenge to the maintenance of pulmonary gas exchange than any other physiologic stressor. This chapter discusses the responses of the healthy respiratory system to exercise with an emphasis on the following problems: What neurochemical mechanisms regulate the ventilatory response to exercise and what are the consequences of this hyperpnea to the work and to the fatigue of the respiratory muscles? What mechanisms underlie the widening of the alveolar to arterial partial pressure of oxygen (PO2) difference during exercise? How do the unique characteristics of the pulmonary circulation determine its response to exercise? How does respiration impact the cardiovascular response to exercise? Under what circumstances might the respiratory system provide a limitation to O2 transport and/or exercise performance? We consider these problems primarily in the healthy, young, normally fit adult, with reference to special cases of the highly trained athlete and to the effects of healthy aging, high altitude hypoxia, and physical training.
EXERCISE HYPERPNEA
In healthy humans, breathing in all physiological states is remarkably well controlled. Accordingly, the partial pressures of oxygen and carbon dioxide, in systemic arterial blood along with its acidity, are regulated precisely throughout mild to moderate exercise.1–4
These relationships are shown in the following alveolar gas equations, where alveolar gas partial pressures are approximately equal to the ratio of the metabolic requirement to alveolar ventilation.
![]()
![]()
where:
PACO2 and PAO2 = alveolar carbon dioxide and oxygen partial pressures (it is assumed PACO2 ≈ arterial PCO2)
![]() and
and ![]() = volumes of carbon dioxide produced and oxygen consumed
= volumes of carbon dioxide produced and oxygen consumed
![]() = alveolar ventilation
= alveolar ventilation
PIO2 = inspired partial pressure of oxygen
K = constant (0.863). This constant allows alveolar gases to be calculated from these equations if ![]() and
and ![]() are expressed in mL/min and
are expressed in mL/min and ![]() in L/min.
in L/min.
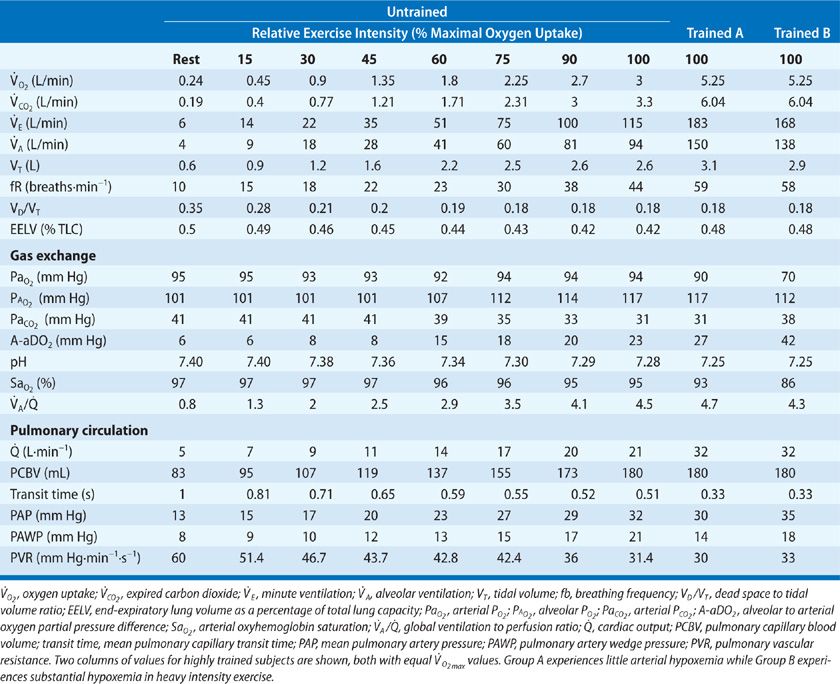
Table 18-1 illustrates the interrelation of these variables, as one goes from rest to exercise. With exercise, there is an increased metabolic rate, with alveolar ventilation increasing to regulate arterial blood gases near resting levels. In health, dead space (VD) increases slightly as intrathoracic airways stretch and dilate with increased tidal volume (VT) – but VT rises out of proportion; thus, VD/VT falls to about one-half its resting value during exercise. In order that PACO2 be precisely controlled, overall minute ventilation (![]() E) during exercise must be regulated in such a fashion so as to compensate both for the increasing CO2 production as well as a reduced VD/VT. Accordingly, the ratio of
E) during exercise must be regulated in such a fashion so as to compensate both for the increasing CO2 production as well as a reduced VD/VT. Accordingly, the ratio of ![]() E to
E to ![]() falls from rest to moderate exercise, whereas the
falls from rest to moderate exercise, whereas the ![]() A:
A:![]() ratio and arterial PCO2 are maintained constant until heavy exercise intensities, during which both ratios rise and PACO2 is reduced. Note the impressive magnitude of ventilatory response required to maintain arterial PCO2 homeostasis during exercise, amounting to a 20-fold increase above resting in the untrained at
ratio and arterial PCO2 are maintained constant until heavy exercise intensities, during which both ratios rise and PACO2 is reduced. Note the impressive magnitude of ventilatory response required to maintain arterial PCO2 homeostasis during exercise, amounting to a 20-fold increase above resting in the untrained at ![]() and 30-fold in the highly trained.
and 30-fold in the highly trained.
TABLE 18-1 Group Mean Values of Healthy Untrained and Trained Subjects Cardiorespiratory Responses to Steady-State Exercise
 REGULATION OF EXERCISE HYPERPNEA
REGULATION OF EXERCISE HYPERPNEA
More than a century of highly innovative research on this question has left us with three major stimuli as the primary regulators of exercise hyperpnea. The schematic diagram shown in Figure 18-1 includes these three potential stimuli in a ventilatory control system which features three components, namely a central rhythm generator/integrator in the medulla, mechanical and chemical feedback and feedforward inputs to this integrator, and control of the distribution of efferent output to muscles of both the upper airway and chest and abdominal wall.
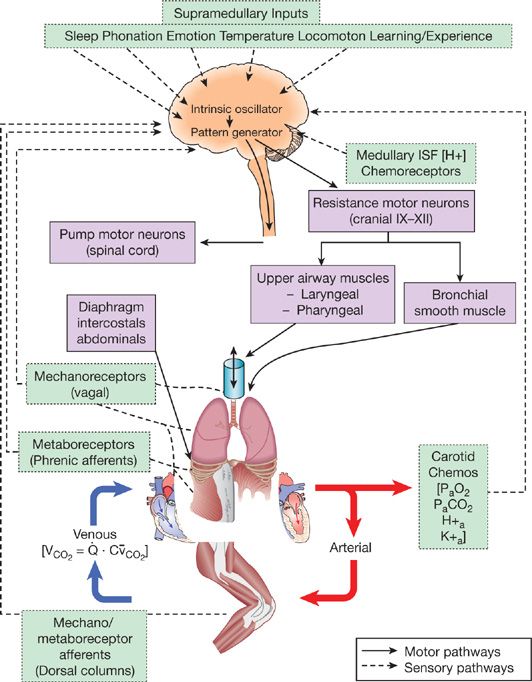
Figure 18-1 Schematic depicting multiple structures contributing to the control of breathing. It is hypothesized that respiratory rhythm originates within a brain stem oscillator which activates brain stem pattern generating neurons that provide for the proper sequential activation of respiratory pump (diaphragm, intercostal, and abdominal) and airway (laryngeal and pharyngeal) muscles. These brain stem neurons receive excitatory and inhibitory input from multiple sources during exercise, including supramedullary central command and mechano/metaboreceptor-initiated spinal afferents from limb and respiratory skeletal muscles. In addition, the brain stem controller neurons receive carotid and intracranial chemoreceptor (RTN, retrotrapezoid nucleus) and vagal mechanoreceptor input critical to meet the appropriate ventilatory and breathing pattern responses to exercise. (Reproduced with permission from Taylor N. Physiological Bases of Human Performance during work and exercise. New York: Churchill Livinstone; 2008.)
 CO2 FLOW
CO2 FLOW
The primary suspects for mediation of the exercise hyperpnea include humoral stimuli in the form of CO2 flow to the lung or the product of blood flow and mixed venous CO2 content. While still controversial (and mysterious) there is compelling evidence in support of a significant fundamental role for this feedback mechanism. When extracorporeal perfusion is used to increase (or reduce) CO2 flow to the lung in a resting animal, alveolar ventilation is changed in proportion to ![]() and an isocapnic hyperpnea (or reduced ventilation) is achieved.5 In human quadriplegic patients, increasing locomotor muscle CO2 production via electrical stimulation of muscle contraction, increases ventilation. Similarly, increasing
and an isocapnic hyperpnea (or reduced ventilation) is achieved.5 In human quadriplegic patients, increasing locomotor muscle CO2 production via electrical stimulation of muscle contraction, increases ventilation. Similarly, increasing ![]() and the respiratory quotient with bicarbonate ingestion in resting humans will increase
and the respiratory quotient with bicarbonate ingestion in resting humans will increase ![]() A in an isocapnic fashion. Furthermore, when sinusoidal exercise regimens are employed at changing frequencies, the ventilatory response follows the change in
A in an isocapnic fashion. Furthermore, when sinusoidal exercise regimens are employed at changing frequencies, the ventilatory response follows the change in ![]() rather than the change in work rate, per se. The case against this purely humoral feedback stimulus in the mediation of exercise hyperpnea is that it has only been tested over a very narrow range of
rather than the change in work rate, per se. The case against this purely humoral feedback stimulus in the mediation of exercise hyperpnea is that it has only been tested over a very narrow range of ![]() near resting levels and the exact nature of the stimulus or its site of action has not been identified. Recently, c-fiber receptors in the lung interstitium have been implicated, responding to an increased transport of plasma water into the lung interstitium, secondary to the effects of both increased venous CO2 content on the osmotic state of blood plasma and increased blood flow on pulmonary capillary pressure.6 We suspect that
near resting levels and the exact nature of the stimulus or its site of action has not been identified. Recently, c-fiber receptors in the lung interstitium have been implicated, responding to an increased transport of plasma water into the lung interstitium, secondary to the effects of both increased venous CO2 content on the osmotic state of blood plasma and increased blood flow on pulmonary capillary pressure.6 We suspect that ![]() plays an important modulatory role in the control of breathing near resting levels, but it is unlikely to provide sufficient drive to be considered as a primary drive to hyperpnea during exercise.7
plays an important modulatory role in the control of breathing near resting levels, but it is unlikely to provide sufficient drive to be considered as a primary drive to hyperpnea during exercise.7
 CENTRAL COMMAND
CENTRAL COMMAND
A purely feedforward input to medullary respiratory controller neurons originates from supramedullary regions of the motor cortex and hypothalamus and operates along synaptic pathways in parallel with the motor control of locomotor muscles. Animal models using electrical or pharmacological stimulation of these supramedullary sites show powerful cardioventilatory responses even in the face of paralyzed locomotor muscles, that is, in the absence of neural feedback.8 Further, in hypnotized humans even the “suggestion” of exercise – while still at rest – elicits exercise-like cardioventilatory responses with coincident PET imaging studies showing an increased blood flow to motor control regions of the cortex.9 On the other hand, the “exercise-like” ventilatory response to electrical stimulation of the limbs shows that feedforward central command is not obligatory to the hyperpnea and a normal ventilatory response to exercise is also observed in decorticate animals, that is, in the absence of key hypothalamic regions of purported cardioventilatory central command.
 MUSCLE AFFERENT FEEDBACK
MUSCLE AFFERENT FEEDBACK
Lightly and unmyelinated afferents from locomotor muscle sensitive to the metabolic milieu, mechanical deformation, and even vascular distension in the muscle, project via the dorsal horn of the spinal cord and then via the nucleus of the solitary tract to the medullary cardiorespiratory controller neurons. When their effect on ventilation is studied in isolation, using electrical stimulation of muscle, a proportionate increase in ![]() E occurs. However, evidence against a role for muscle afferents in the intact human include the failure of imposed vascular occlusion causing accumulation of muscle metabolites to augment ventilation during recovery from exercise, the failure of spinal cord lesioning to alter the ventilatory response to muscle stimulation, or of epidural anesthesia to reduce the cardioventilatory response to rhythmic exercise. On the other hand, if afferent blockade techniques are employed – such as intrathecal fentanyl which blocks only muscle afferents and leaves efferent motor pathways intact, hypoventilation occurs throughout mild and moderate exercise – uncovering a significant obligatory role for muscle afferents in the steady-state exercise hyperpnea (see Fig. 18-2).10,11 This inhibitory effect of muscle afferent blockade on exercise hyperpnea has also been shown in patients with COPD and CHF.12,13 Interestingly, in COPD patients muscle afferent blockade during exercise reduced primarily VD ventilation, suppressed dyspneic sensations, and substantially improved exercise performance.12
E occurs. However, evidence against a role for muscle afferents in the intact human include the failure of imposed vascular occlusion causing accumulation of muscle metabolites to augment ventilation during recovery from exercise, the failure of spinal cord lesioning to alter the ventilatory response to muscle stimulation, or of epidural anesthesia to reduce the cardioventilatory response to rhythmic exercise. On the other hand, if afferent blockade techniques are employed – such as intrathecal fentanyl which blocks only muscle afferents and leaves efferent motor pathways intact, hypoventilation occurs throughout mild and moderate exercise – uncovering a significant obligatory role for muscle afferents in the steady-state exercise hyperpnea (see Fig. 18-2).10,11 This inhibitory effect of muscle afferent blockade on exercise hyperpnea has also been shown in patients with COPD and CHF.12,13 Interestingly, in COPD patients muscle afferent blockade during exercise reduced primarily VD ventilation, suppressed dyspneic sensations, and substantially improved exercise performance.12
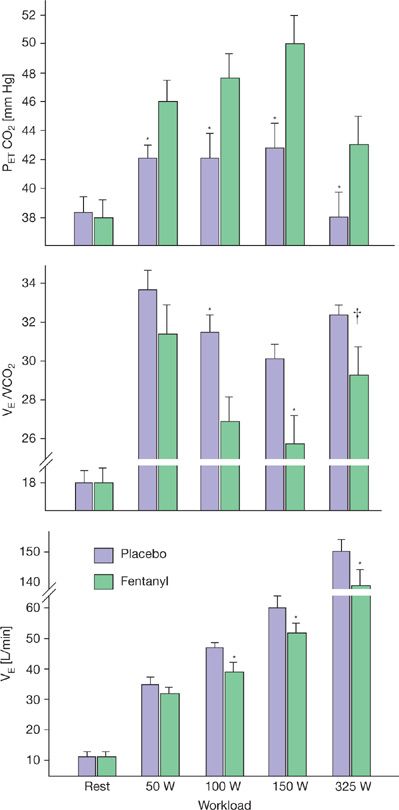
Figure 18-2 Effect of blockage of μ-opioid sensitive type III–IV muscle afferents via intrathecal fentanyl on the steady-state ventilatory response to 3 minutes of cycling exercise at each of four work rates. (*p < 0.05, †p < 0.08). The fentanyl-induced hypoventilation was due to a reduced breathing frequency. Heart rate, mean arterial blood pressure, and ![]() E were significantly reduced at each work rate. Taking into account the reduced exercise
E were significantly reduced at each work rate. Taking into account the reduced exercise ![]() E with fentanyl plus the ventilatory equivalent of the concomitant rise in PETCO2, it is estimated that the partial blockage of muscle afferents accounted for 47%, 45%, and 15% of the total exercise hyperpnea at 100, 150, and 325 W, respectively. (Modified with permission from Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Group III and IV muscle afferents contribute to ventilatory and cardiovascular response to rhythmic exercise in humans. J Appl Physiol. 2010;109(4):966–976.)
E with fentanyl plus the ventilatory equivalent of the concomitant rise in PETCO2, it is estimated that the partial blockage of muscle afferents accounted for 47%, 45%, and 15% of the total exercise hyperpnea at 100, 150, and 325 W, respectively. (Modified with permission from Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Group III and IV muscle afferents contribute to ventilatory and cardiovascular response to rhythmic exercise in humans. J Appl Physiol. 2010;109(4):966–976.)
In summary, the dilemma of exercise hyperpnea is that findings using isolation of each of these three stimuli support a significant contributory role for each of these mechanisms to the isocapnic hyperpnea of moderate intensity exercise—but there is contradictory evidence against an obligatory major role for any of them. Accordingly, most models emphasize the powerful redundancy of the hyperpnea mechanisms operating under steady-state conditions of exercise or emphasize the importance of compensatory feedback from carotid and central chemoreceptors if blockade of any of these primary mechanisms was sufficient to cause transient CO2 retention. There are also suggestions that feedback mechanisms are likely of little consequence; rather, the ventilatory response to exercise depends critically on a “stored memory” of the appropriate ventilatory response by the motor cortex as a result of repetitive trial and error during maturation.14 We hypothesize – with limited direct evidence – that each of these three mechanisms have an important obligatory role, with CO2 flow to the lung providing the essential underpinning to ventilatory control in all physiologic states and the interaction of feedforward (central command) and feedback (from muscle afferents) mechanisms providing the primary “exercise” stimulus. The proof for this awaits the appropriate experimental tools – such as was done with the use of opiate receptor agonists (see Fig. 18-2) – to test a specific mechanism without altering the remaining elements of the control system.
Finally, we need to emphasize that this hypothesis applies only to the isocapnic hyperpnea achieved in mild to moderate steady-state levels of exercise. For the hyperventilatory response to heavier intensity exercise we need to add additional mechanisms: an important input from carotid chemoreceptors responding to the changing hydrogen ion, potassium, norepinephrine, temperature, etc. of the arterial blood induced by heavy intensity exercise; the additional powerful inputs from central command responding to the need to recruit more motor units in the presence of fatiguing locomotor muscles; and muscle afferents responding to accumulating ionic changes in the muscle interstitium. Even an obligatory role for carotid chemoreceptors and the hyperventilatory response to heavy exercise has been challenged by the fact that preventing the blood-borne acidity during heavy exercise (via depleting muscle glycogen and preventing acid production) did not prevent the hyperventilatory response.7 Again multiple, redundant mechanisms are apparently at play. This hyperventilatory response is extremely important in partially compensating for the metabolic acidosis incurred with increasing lactic acid levels in heavy exercise and also for raising alveolar PO2 to maintain arterial PO2 in the face of a widening alveolar to arterial PO2 difference.15
 BREATHING PATTERN DURING EXERCISE
BREATHING PATTERN DURING EXERCISE
During low to moderate intensity exercise both VT and breathing frequency (fb) increase roughly in proportion to intensity, while at higher intensities VT attains a plateau and further increases in ![]() E are accomplished by increases in fb alone (see Fig. 18-3). The increase in fb is accomplished by decreases in both inspiratory time (TI) and expiratory time (TE). However the ratio of TI to total breath cycle duration (TTOT), known as the duty cycle (TI/TTOT), increases only slightly during exercise (∼0.40 at rest to ∼0.50 during high-intensity exercise). The fact that the duty cycle remains low is important and beneficial, because prolonged diaphragmatic contractions hinder blood flow to this muscle and may precipitate excessive diaphragmatic fatigue.16
E are accomplished by increases in fb alone (see Fig. 18-3). The increase in fb is accomplished by decreases in both inspiratory time (TI) and expiratory time (TE). However the ratio of TI to total breath cycle duration (TTOT), known as the duty cycle (TI/TTOT), increases only slightly during exercise (∼0.40 at rest to ∼0.50 during high-intensity exercise). The fact that the duty cycle remains low is important and beneficial, because prolonged diaphragmatic contractions hinder blood flow to this muscle and may precipitate excessive diaphragmatic fatigue.16
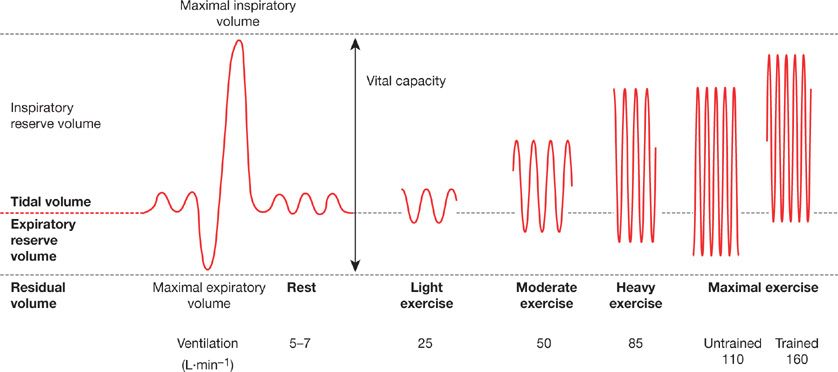
Figure 18-3 Changes in breathing pattern during exercise. The spirograph on the left is from a resting subject showing normal tidal volume, maximum expiration to residual lung volume, then maximal inspiration to total lung capacity. With light to heavy exercise (in both untrained and highly trained subjects) the increase in ventilation is achieved by increasing breathing frequency and tidal volume. Tidal volume increases by encroaching on the expiratory and inspiratory reserve volumes. The reduced end-expiratory lung volume is maintained at maximal exercise in the normally fit subject (maximal oxygen uptake [![]() ] 45 mL kg–1 L min–1). In the trained subject (
] 45 mL kg–1 L min–1). In the trained subject (![]() = 75 mL kg–1 L min–1), ventilation, breathing frequency, and tidal volume are all higher and maximal exercise end-expiratory lung volume is increased to near resting values due to expiratory flow limitations. (Reproduced with permission from Farrell PA, Joyner MJ, Caiozxo VJ.ACSM’s Advanced Exercise Physiology, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.)
= 75 mL kg–1 L min–1), ventilation, breathing frequency, and tidal volume are all higher and maximal exercise end-expiratory lung volume is increased to near resting values due to expiratory flow limitations. (Reproduced with permission from Farrell PA, Joyner MJ, Caiozxo VJ.ACSM’s Advanced Exercise Physiology, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.)
The increase in VT at the onset of exercise is accomplished by both an increase in end-inspiratory lung volume (EILV) and a decrease in end-expiratory lung volume (EELV). However, as exercise intensity increases, EILV does not normally increase beyond 85% to 90% TLC. Beyond this point lung compliance decreases markedly and the respiratory pressure production required for a given change in volume is very large. This inefficiency at high operating lung volumes leads to neuromechanical uncoupling in that a mismatch develops between the required “effort” to inspire and the actual volume of air inhaled. This hyperinflation (secondary to expiratory flow limitation) underlies much of exertional dyspnea, that is, “unsatisfied inspiratory effort” in COPD patients.17 In health, EELV decreases at the onset of all levels of exercise due to active recruitment of the expiratory muscles, and its decrease is roughly proportional to exercise intensity.18 The drop in EELV maintains operating lung volumes within the linear portion of the pressure–volume relationship, which minimizes the reduction in respiratory system compliance and associated dyspnea that develops at high lung volumes. The reduced EELV also serves to lengthen the diaphragm and place it in a more optimal range of its length–tension relationship. Thus the maximum dynamic capacity of the inspiratory muscles for force production is improved during tidal breathing and they are required to produce only about one-half of their capacity for force production at maximum exercise in the untrained subject (see Fig. 18-3).19 A reduced EELV also reduces inspiratory muscle work during the ensuing inspiration due to outward recoil of the rib cage at the onset of inspiration.
Tidal exercise flow–volume loops plotted within the maximal volitional flow–volume envelope provide a simple and useful method to analyze alterations in flow rates, VT, and operating lung volumes during exercise (Fig. 18-4). In normal, healthy untrained young adult humans the maximal attainable flow rates at any given lung volume are usually much greater than the spontaneous tidal flow rates reached during exercise of all intensities. Thus, as shown in Figure 18-4 (at ![]() Emax in the 100–120 L/min range) there is usually a large reserve for increasing
Emax in the 100–120 L/min range) there is usually a large reserve for increasing ![]() E even at maximal exercise.
E even at maximal exercise.
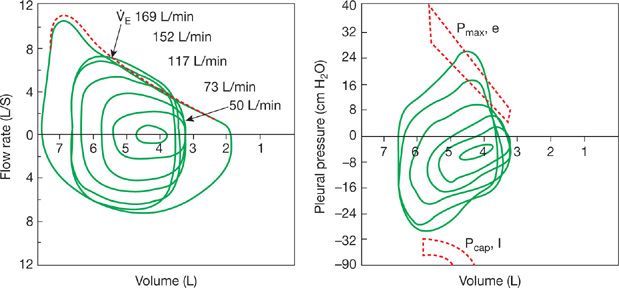
Figure 18-4 Flow–volume and pressure–volume relationships in a young healthy adult, at rest and during exercise. The maximal (outer envelope) flow–volume relationship is obtained via maximal volitional inspiratory and expiratory efforts, before (solid line) and immediately following exercise (broken line). For the pressure–volume relationships, only tidal breaths from rest through to maximal exercise are shown. In addition, the maximum inspiratory pleural pressures (Pcap, I) are shown at the specific peak volume and flows achieved during tidal breathing in heavy exercise. For minute ventilations up to 115 L min–1, approximating peak exercise in an untrained adult, the inspiratory muscles are activated to only ∼40% to 50% of capacity. The more highly trained subject is shown achieving ventilations >150 L min–1 at higher metabolic rates. Under these conditions, the tidal flow–volume loop often encroaches on the maximum flow–volume envelope, end-expiratory lung volume rises, and the inspiratory muscles approach 90% of their dynamic capacity for force output and shortening velocity. The broken area on the expiratory side indicates expiratory pressures for any given lung volume (Pmax, e), beyond which extraexpiratory muscle effort will not produce a higher flow. In almost all instances up to ventilations of ∼150 L min–1, this critical expiratory pressure is not exceeded but it is exceeded slightly in the highly trained athlete at maximum exercise. (Modified with permission from Johnson BD, Saupe KW, Dempsey JA. Mechanical constraints on exercise hyperpnea in endurance athletes. J Appl Physiol. 1992;73(3):874–886.)
CONTROL OF AIRWAY CALIBER DURING EXERCISE
 UPPER AIRWAY CALIBER
UPPER AIRWAY CALIBER
The upper airway comprises the nose, mouth, pharynx, and larynx, and provides the majority of resistance to airflow at rest and during exercise. In addition, each region of the upper airway has the potential to independently contribute to any alterations in airway resistance during exercise. The work required to produce the large increases in airflow during exercise would become excessive during even low-intensity exercise if several mechanisms were not in place to reduce resistance to airflow in the upper airway during exercise.
First, the route of airflow switches from predominately nasal to oronasal breathing when ![]() E reaches approximately 30 L/min.20 Second, nasal resistance decreases during exercise in an intensity- and duration-dependent manner secondary to sympathetically mediated vasoconstriction of the nasal mucosal vasculature.21 Third, the nasal dilator muscles and presumably the skeletal muscles of the pharyngeal and laryngeal regions contract in phase with, but slightly preceding, inspiratory pump muscle recruitment, and this drive to the upper airway muscles is increased at increasing
E reaches approximately 30 L/min.20 Second, nasal resistance decreases during exercise in an intensity- and duration-dependent manner secondary to sympathetically mediated vasoconstriction of the nasal mucosal vasculature.21 Third, the nasal dilator muscles and presumably the skeletal muscles of the pharyngeal and laryngeal regions contract in phase with, but slightly preceding, inspiratory pump muscle recruitment, and this drive to the upper airway muscles is increased at increasing ![]() E, resulting in decreased resistance and a larger diameter, stiffer, less collapsible upper airway.22 Finally, the glottic narrowing that normally occurs during expiration is attenuated during exercise due to laryngeal abductor muscle activation, in addition to a widened mean glottic aperture throughout the respiratory cycle.23 Thus, the work required to produce the increased airflow that occurs during whole-body exercise is minimized by a variety of adjustments that occur in the upper airway, all of which act to decrease resistance to airflow.
E, resulting in decreased resistance and a larger diameter, stiffer, less collapsible upper airway.22 Finally, the glottic narrowing that normally occurs during expiration is attenuated during exercise due to laryngeal abductor muscle activation, in addition to a widened mean glottic aperture throughout the respiratory cycle.23 Thus, the work required to produce the increased airflow that occurs during whole-body exercise is minimized by a variety of adjustments that occur in the upper airway, all of which act to decrease resistance to airflow.
 BRONCHIAL CALIBER
BRONCHIAL CALIBER
Bronchial dilation in response to exercise has been well documented in healthy humans.24 Furthermore, this bronchodilator influence occurs at exercise onset and is very powerful, as evidenced by the prevention of an increase in pulmonary resistance during exercise after histamine inhalation in asthmatic subjects who exhibited large increases in resistance during histamine inhalation at rest.25 In addition, forced expiratory volume in 1 second (FEV1) and the maximum volitional flow:volume envelope increase immediately after exercise in both normal26 and asthmatic subjects.27 There are several potential mechanisms contributing to the bronchodilator effect of exercise, including neural, mechanical, and locally released mediator mechanisms.
A primary component of the exercise-induced increase in airway caliber is withdrawal of vagal parasympathetic tone to the airways that occurs at the immediate onset of exercise, resulting in bronchial smooth muscle relaxation.24 The withdrawal of cholinergic tone is thought to be mediated in part by the stimulation of muscle mechano- and chemosensitive afferents (i.e., the same muscle afferents believed to be involved in the pressor and ventilatory responses to exercise) (see Fig. 18-2).28 Increased lung stretch and activation of slowly adapting pulmonary stretch receptors (that occurs as EILV is increased during exercise) may contribute to this withdrawal of vagal tone.
Mechanical influences may also play a substantial role in increasing airway caliber during exercise. The airways are tethered open by the lung parenchyma, and the increase in EILV and operating lung volumes during exercise will enlarge airway diameter simply as a result of this airway–parenchymal interdependence. Further, the increased outward radial force exerted by the parenchyma on the airways during exercise may induce bronchodilation by a separate mechanism operating at the level of the crossbridges of bronchial smooth muscle.29 Airway stretch modulates bronchial smooth muscle crossbridge formation, resulting in decreased bronchial smooth muscle force and stiffness and relaxation of airway smooth muscle. Finally, airway mast cells, macrophages, neutrophils, eosinophils, epithelial cells, and smooth muscle cells all have the potential to release a variety of chemical mediators that may alter airway caliber.
WORK, METABOLIC AND CIRCULATORY COST OF BREATHING
During exercise the inspiratory and expiratory muscles perform work on the lung, the abdominal wall, and the rib cage. The work on the lung is composed of elastic work – a function of both the VT and lung compliance and flow resistive work – a function of airway caliber and flow rate. As the diaphragm descends during inspiration, work is performed on the abdominal wall—a function of abdominal wall compliance. This work maximally contributes about 25% to the total work of breathing during exercise—but in the presence of expiratory flow limitation abdominal muscle tension will persist well into the inspiration which will markedly increase work done by the diaphragm on the abdominal compartment. As shown in Figure 18-5 the total work of breathing rises linearly with the isocapnic hyperpnea of mild and moderate intensity exercise and then rises alinearly with the hyperventilation of heavy intensity exercise.
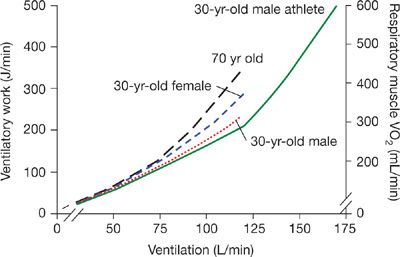
Figure 18-5 Ventilatory work and respiratory muscle ![]() during exercise of increasing intensity plotted as a function of
during exercise of increasing intensity plotted as a function of ![]() E for sedentary men, active young females, and trained young and older men. In young adult males the O2 cost of exercise hyperpnea was determined by having subjects mimic the pressure–volume loop, breathing frequency, duty cycle, and ventilation they experienced during submaximal and maximal exercise and measuring the change in
E for sedentary men, active young females, and trained young and older men. In young adult males the O2 cost of exercise hyperpnea was determined by having subjects mimic the pressure–volume loop, breathing frequency, duty cycle, and ventilation they experienced during submaximal and maximal exercise and measuring the change in ![]() from resting eupnea.108,109 (Reproduced with permission from Harms CA, Dempsey JA. Cardiovascular consequences of exercise hyperpnea. Exerc Sport Sci Rev. 1999;27:37–62.)
from resting eupnea.108,109 (Reproduced with permission from Harms CA, Dempsey JA. Cardiovascular consequences of exercise hyperpnea. Exerc Sport Sci Rev. 1999;27:37–62.)
This severalfold increase in respiratory muscle work requires increases in respiratory muscle ![]() and blood flow. Thus, at maximal
and blood flow. Thus, at maximal ![]() (∼45 mL/kg/min) and cardiac output (∼20 L/min) and
(∼45 mL/kg/min) and cardiac output (∼20 L/min) and ![]() E (100–120 L/min), about 8% to 10% of
E (100–120 L/min), about 8% to 10% of ![]() and cardiac output are devoted to breathing in the untrained human and up to 14% to 16% of
and cardiac output are devoted to breathing in the untrained human and up to 14% to 16% of ![]() and cardiac output in the highly trained (
and cardiac output in the highly trained (![]() ∼65 mL/kg,
∼65 mL/kg, ![]() E >150 L/min, and CO ∼30 L/min—see Table 18-1). Respiratory muscle blood flow, was determined directly by using distribution of infused microspheres in exercising animals30 and indirectly in humans by the reduction in total cardiac output and increased blood flow to the exercising limb muscles measured when respiratory muscle work was reduced during maximal exercise using a mechanical ventilator (see Fig. 18-6).31,32
E >150 L/min, and CO ∼30 L/min—see Table 18-1). Respiratory muscle blood flow, was determined directly by using distribution of infused microspheres in exercising animals30 and indirectly in humans by the reduction in total cardiac output and increased blood flow to the exercising limb muscles measured when respiratory muscle work was reduced during maximal exercise using a mechanical ventilator (see Fig. 18-6).31,32
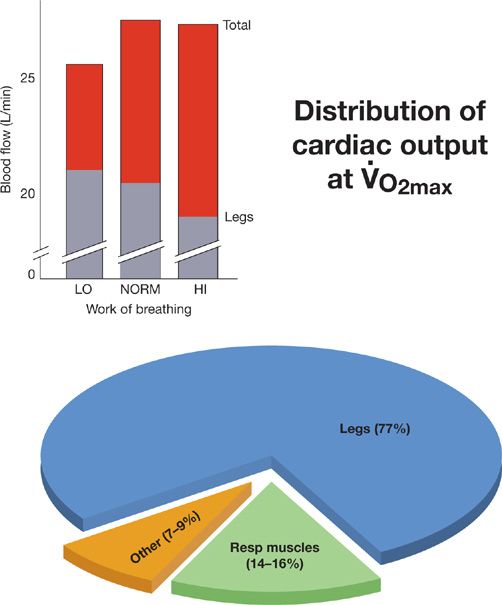
Figure 18-6 Effects of respiratory muscle work during exercise on cardiac output and its distribution in highly fit adult male subjects cycling at ![]() (
(![]() = 65 mL/kg/min, cardiac output = 28 L/min), “LO, NORM, HI” refer to the relative level of the work of breathing present during heavy intensity exercise under normal physiologic conditions, with added resistive loads (“HI”) and during unloading of the respiratory muscles via mechanical ventilation (“LO”). The top of each bar indicates the total cardiac output and each bar is divided into blood flow to the limbs and to the rest of the body. The estimated distribution of blood flow to the limb locomotor and to the respiratory muscles is shown in the pie chart. These estimates come from three sources: 1. The oxygen cost of breathing at maximum exercise108; 2. Measurements based on microsphere distribution to the respiratory muscles during maximum exercise in the equine110; and 3. The change in cardiac output and in limb muscle blood flow determined in response to unloading of the respiratory muscles during maximum exercise.31,32 These effects of respiratory muscle unloading at maximum exercise on limb blood flow and on total cardiac output are shown in the insert. Note that with reduced respiratory muscle work, that is, unloading, the total cardiac output falls and the limb muscle blood flow (and vascular conductance) rises; whereas with respiratory muscle loading and increased work of breathing at maximum exercise the maximum cardiac output remains unchanged whereas the limb blood flow (and vascular conductance) is reduced. (Reproduced with permission from Farrell PA, Joyner MJ, Caiozxo VJ. ACSM’s Advanced Exercise Physiology, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.)
= 65 mL/kg/min, cardiac output = 28 L/min), “LO, NORM, HI” refer to the relative level of the work of breathing present during heavy intensity exercise under normal physiologic conditions, with added resistive loads (“HI”) and during unloading of the respiratory muscles via mechanical ventilation (“LO”). The top of each bar indicates the total cardiac output and each bar is divided into blood flow to the limbs and to the rest of the body. The estimated distribution of blood flow to the limb locomotor and to the respiratory muscles is shown in the pie chart. These estimates come from three sources: 1. The oxygen cost of breathing at maximum exercise108; 2. Measurements based on microsphere distribution to the respiratory muscles during maximum exercise in the equine110; and 3. The change in cardiac output and in limb muscle blood flow determined in response to unloading of the respiratory muscles during maximum exercise.31,32 These effects of respiratory muscle unloading at maximum exercise on limb blood flow and on total cardiac output are shown in the insert. Note that with reduced respiratory muscle work, that is, unloading, the total cardiac output falls and the limb muscle blood flow (and vascular conductance) rises; whereas with respiratory muscle loading and increased work of breathing at maximum exercise the maximum cardiac output remains unchanged whereas the limb blood flow (and vascular conductance) is reduced. (Reproduced with permission from Farrell PA, Joyner MJ, Caiozxo VJ. ACSM’s Advanced Exercise Physiology, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.)
Several lines of evidence are available in support of the concept that during heavy intensity exercise a metaboreflex is triggered from the diaphragm which travels in phrenic nerve afferent fibers to increase efferent vasoconstrictor outflow from sympathetic neurons in the medulla. For example, studies in healthy humans using muscle microneurography and Doppler blood flow measurements of limb muscle blood flow demonstrated increases in muscle sympathetic nerve activity together with reduced limb vascular conductance and blood flow in the face of inspiratory or expiratory muscle fatigue induced by volitional hyperpnea against increased airway resistive loads.33
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


