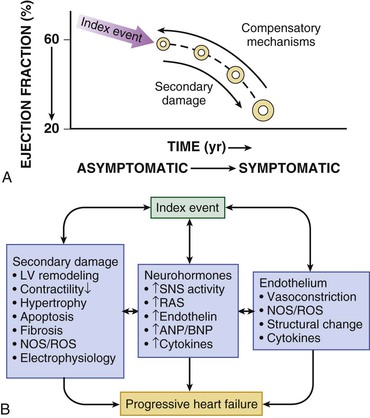
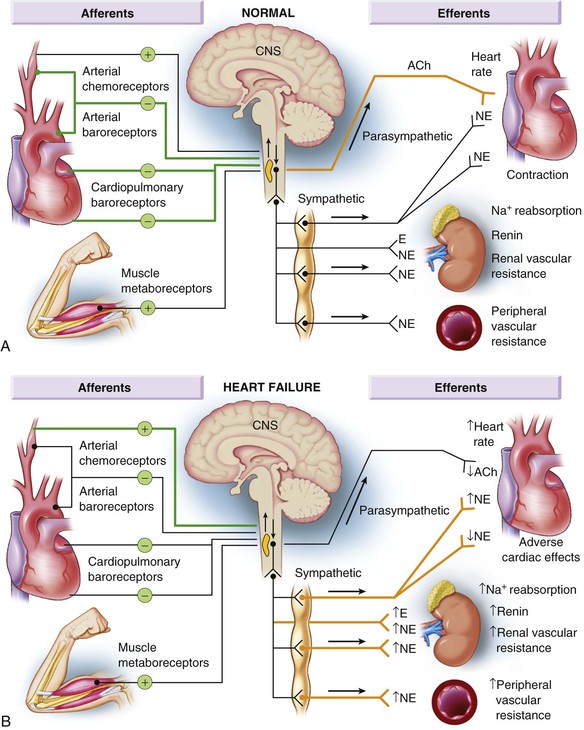
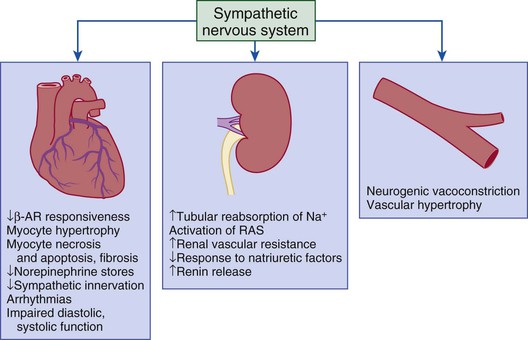
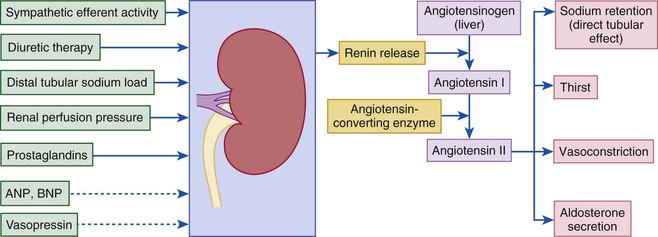
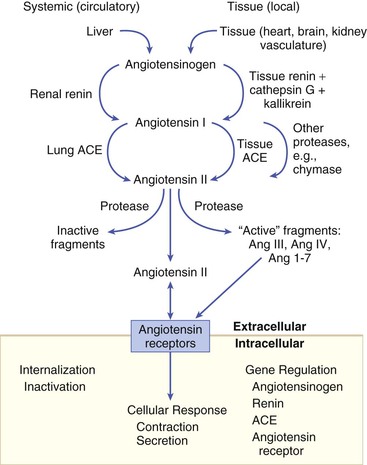
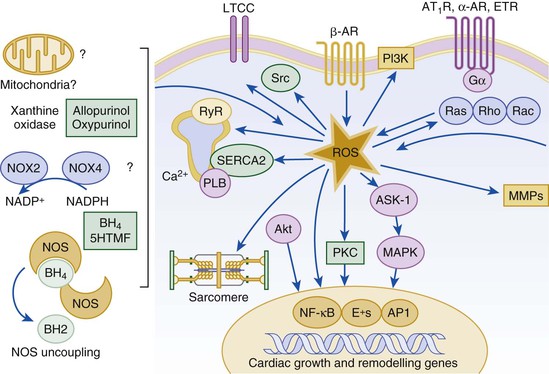
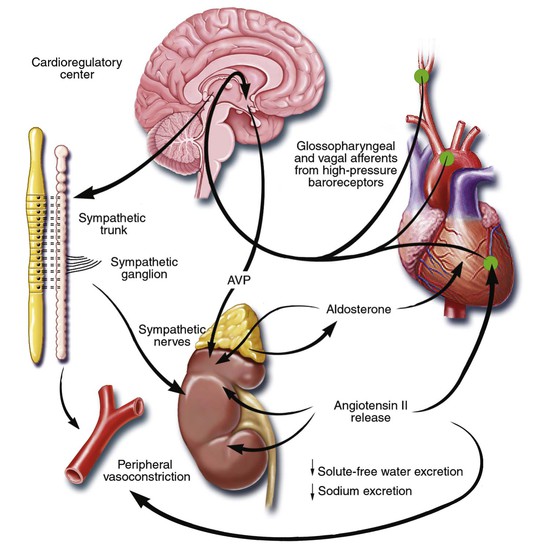
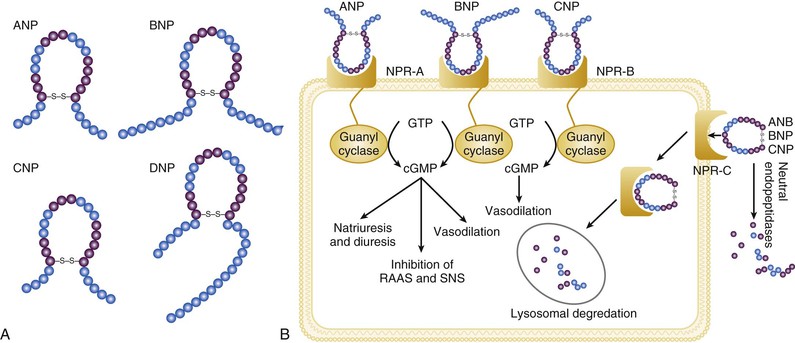
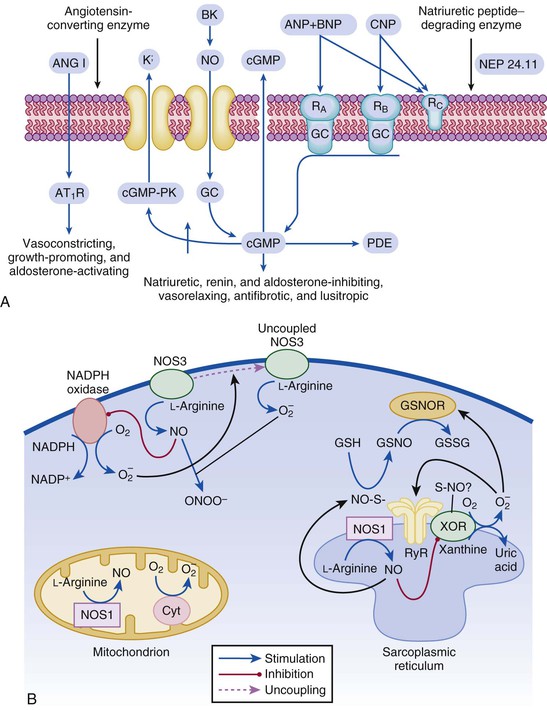
Gerd Hasenfuss, Douglas L. Mann Despite repeated attempts to discover a unique pathophysiologic mechanism that precisely explains the clinical syndrome of heart failure, no single conceptual paradigm has withstood the test of time. Although clinicians initially viewed heart failure as a problem of excessive salt and water retention that was caused by abnormalities of renal blood flow (the so-called cardiorenal model) and/or abnormal pumping capacity of the heart (the cardiocirculatory or hemodynamic model),1 these models do not adequately explain the relentless disease progression that occurs in this syndrome. This chapter focuses on the molecular and cellular changes that underlie heart failure with depressed systolic function, with an emphasis on the role of neurohormonal activation and left ventricular (LV) remodeling as the primary determinants for disease progression in heart failure. The hemodynamic, contractile, and wall motion disorders in heart failure are discussed in the chapters on echocardiography (see Chapter 14), cardiac catheterization (see Chapter 19), radionuclide imaging (see Chapter 16), and clinical assessment of the patient with heart failure (see Chapter 23). The pathogenesis of heart failure with a normal ejection fraction is discussed elsewhere in this book (see Chapter 27). As shown in Figure 22-1A, heart failure may be viewed as a progressive disorder that is initiated after an index event either damages the heart muscle, with a resultant loss of functioning cardiac myocytes or, alternatively, disrupts the ability of the myocardium to generate force, thereby preventing the heart from contracting normally. This index event may have an abrupt onset, as in the case of a myocardial infarction; it may have a gradual or insidious onset, as in the case of hemodynamic pressure or volume overloading, or it may be hereditary, as in the case of many of the genetic cardiomyopathies. Regardless of the nature of the inciting event, the feature that is common to each of these index events is that they all, in some manner, produce a decline in pumping capacity of the heart. In most instances, patients will remain asymptomatic or minimally symptomatic after the initial decline in pumping capacity of the heart, or symptoms develop only after the dysfunction has been present for some time. Although the precise reasons why patients with LV dysfunction remain asymptomatic have not been established with certainty, one potential explanation is that a number of compensatory mechanisms that become activated in the setting of cardiac injury or depressed cardiac output appear to modulate LV function within a physiologic/homeostatic range, such that the patient’s functional capacity is preserved or is depressed only minimally. With progression to symptomatic heart failure, however, the sustained activation of neurohormonal and cytokine systems leads to a series of end-organ changes within the myocardium referred to collectively as LV remodeling. As discussed further on, LV remodeling is sufficient to lead to disease progression in heart failure independent of the neurohormonal status of the patient. A growing body of experimental and clinical evidence suggests that heart failure progresses as a result of the overexpression of biologically active molecules that are capable of exerting deleterious effects on the heart and circulation (Fig. 22-1B).1 The portfolio of compensatory mechanisms that have been described thus far includes activation of the adrenergic nervous systemic system and the renin-angiotensin system (RAS), which are responsible for maintaining cardiac output through increased retention of salt and water; peripheral arterial vasconstriction and increased contractility; and inflammatory mediators that are responsible for cardiac repair and remodeling. It bears emphasis that neurohormone is largely a historical term, reflecting the original observation that many of the molecules that were elaborated in heart failure were produced by the neuroendocrine system and thus acted on the heart in an endocrine manner. It has since become apparent, however, that a great many of the so-called classical neurohormones such as norepinephrine (NE) and angiotensin II are synthesized directly within the myocardium by myocytes and thus act in an autocrine and paracrine manner. Nonetheless, the important unifying concept that arises from the neurohormonal model is that the overexpression of portfolios of biologically active molecules contributes to disease progression by virtue of the deleterious effects these molecules exert on the heart and circulation. The decrease in cardiac output in heart failure activates a series of compensatory adaptations that are intended to maintain cardiovascular homeostasis. One of the most important adaptations is activation of the sympathetic (adrenergic) nervous system, which occurs early in the course of heart failure. Activation of the sympathetic nervous system in heart failure is accompanied by a concomitant withdrawal of parasympathetic tone (Fig. e22-1 As a result of the increase in sympathetic tone, there is an increase in circulating levels of NE, a potent adrenergic neurotransmitter. The elevated levels of circulating NE result from a combination of increased release of NE from adrenergic nerve endings and its consequent “spillover” into the plasma, as well as reduced uptake of NE by adrenergic nerve endings. In patients with advanced heart failure, the circulating levels of NE in resting patients are two to three times those found in normal subjects. Indeed, plasma levels of NE predict mortality in patients with heart failure. Whereas the normal heart usually extracts NE from the arterial blood, in patients with moderate heart failure the coronary sinus NE concentration exceeds the arterial concentration, indicating increased adrenergic stimulation of the heart. However, as heart failure progresses there is a significant decrease in the myocardial concentration of NE. The mechanism responsible for cardiac NE depletion in severe heart failure is not clear and may relate to an “exhaustion” phenomenon resulting from the prolonged adrenergic activation of the cardiac adrenergic nerves in heart failure. In addition, there is decreased activity of myocardial tyrosine hydroxylase, which is the rate-limiting enzyme in the synthesis of NE. In patients with cardiomyopathy, iodine-131–labeled metaiodobenzylguanidine (MIBG), a radiopharmaceutical that is taken up by adrenergic nerve endings, is not taken up normally, suggesting that NE reuptake also islso impaired in heart failure. Increased sympathetic activation of the beta1-adrenergic receptor results in increased heart rate and force of myocardial contraction, with a resultant increase in cardiac output (see Chapter 21). In addition, the heightened activity of the adrenergic nervous system leads to stimulation of myocardial alpha1-adrenergic receptors, which elicits a modest positive inotropic effect, as well as peripheral arterial vasoconstriction (Fig. 22-2). Although NE enhances both contraction and relaxation and maintains blood pressure, myocardial energy requirements are augmented, which can intensify ischemia when myocardial O2 delivery is restricted. The augmented adrenergic outflow from the central nervous system also may trigger ventricular tachycardia or even sudden cardiac death, particularly in the presence of myocardial ischemia. Thus activation of the sympathetic nervous system provides short-term support that has the potential to become maladaptive over the long term (see Fig. 22-2). Moreover, increasing evidence suggests that apart from the deleterious effects of sympathetic activation, parasympathetic withdrawal also may contribute to the pathogenesis of heart failure. Withdrawal of parasympathetic nerve stimulation has been associated with decreased nitric oxide (NO) levels, increased inflammation, increased sympathetic activity and worsening LV remodeling. Two ongoing clinical trials, INOVATE-HF (Increase of Vagal Tone in CHF) (NCT01303718) and NECTAR-HF (Neural Cardiac Therapy for Heart Failure Study) (NCT01385176), are examining the effects of vagal nerve stimulation on LV structure and clinical outcomes in patients with New York Heart Association (NYHA) class III heart failure. In contrast with the sympathetic nervous system, the components of the RAS are activated comparatively later in heart failure. The presumptive mechanisms for RAS activation in heart failure include renal hypoperfusion, decreased filtered sodium reaching the macula densa in the distal tubule, and increased sympathetic stimulation of the kidney, leading to increased renin release from jutaglomerular apparatus (Fig. 22-3). As shown in Figure 22-4, renin cleaves four amino acids from circulating angiotensinogen, which is synthesized in the liver, to form the biologically inactive decapeptide angiotensin I. Angiotensin-converting enzyme (ACE) cleaves two amino acids from angiotensin I to form the biologically active octapeptide(1-8) angiotensin II. Most ACE activity (approaching 90%) in the body is found in tissues; the remaining 10% is found in a soluble (non–membrane-bound) form in the interstitium of the heart and vessel wall. The importance of tissue ACE activity in heart failure is suggested by the observation that ACE messenger RNA (mRNA) and ACE-binding sites and ACE activity are increased in explanted human hearts.3 Angiotensin II also can be synthesized using renin-independent pathways through the enzymatic conversion of angiotensinogen to angiotensin I by kallikrein and cathepsin G (see Fig. 22-4). The tissue production of angiotensin II also may occur along ACE-independent pathways, through the activation of chymase. This latter pathway may be of major importance in the myocardium, particularly when the levels of renin and angiotensin I are increased by the use of ACE inhibitors. Angiotensin II itself can undergo further proteolysis to generate three biologically active fragments: angiotensin III2–8 and angiotensin IV,3–8 which promote vasconstriction,4 and angiotensin,1–7 which may act to counteract the deleterious effects of angiotensin II on endothelial function. Angiotensin II exerts its effects by binding to two G protein–coupled receptors, the angiotensin type 1 (AT1) and angiotensin type 2 (AT2) receptors. The predominant angiotensin receptor in the vasculature is the AT1 receptor. Although both the AT1 and AT2 receptor subtypes are present in human myocardium, the AT2 receptor predominates in a 2:1 molar ratio. Cellular localization of the AT1 receptor in the heart is most abundant in nerves distributed in the myocardium, whereas the AT2 receptor is localized more specifically in fibroblasts and the interstitium. Activation of the AT1 receptor leads to vasoconstriction, cell growth, aldosterone secretion, and catecholamine release, whereas activation of the AT2 receptor leads to vasodilation, inhibition of cell growth, natriuresis, and bradykinin release. Studies have shown that the AT1 receptor and mRNA levels are downregulated in failing human hearts, whereas AT2 receptor density is increased or unchanged, so that the ratio of AT1 to AT2 receptors decreases.4 Angiotensin II has several important actions that are critical to maintaining short-term circulatory homeostasis (see further on). The sustained expression of angiotensin II is maladaptive, however, leading to fibrosis of the heart, kidneys, and other organs. Angiotensin II can also lead to worsening neurohormonal activation by enhancing the release of NE from sympathetic nerve endings, as well as stimulating the zona glomerulosa of the adrenal cortex to produce aldosterone. Analogous to angiotensin II, aldosterone provides short-term support to the circulation by promoting the reabsorption of sodium in exchange for potassium, in the distal segments of the nephron. However, the sustained expression of aldosterone may exert harmful effects by provoking hypertrophy and fibrosis within the vasculature and the myocardium, contributing to reduced vascular compliance and increased ventricular stiffness. In addition, aldosterone provokes endothelial cell dysfunction, baroreceptor dysfunction, and inhibition of NE uptake, any or all of which may lead to worsening of heart failure. The mechanism of action of aldosterone in the cardiovascular system appears to involve oxidative stress, with resultant inflammation in target tissue. Oxidative Stress. Reactive oxygen species (ROS) are a normal byproduct of aerobic metabolism. In the heart, the potential sources for ROS include the mitochondria, xanthine oxidase, and nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase (Fig. 22-5). ROS can modulate the activity of a variety of intracellular proteins and signaling pathways, including essential proteins involved in myocardial excitation-contraction coupling, such as ion channels, sarcoplasmic reticulum (SR) calcium release channels, and myofilament proteins, as well as signaling pathways that are coupled to myocyte growth.5 “Oxidative stress” occurs when the production of ROS exceeds the buffering capacity of antioxidant defense systems, leading to an excess of ROS within the cell. Substantial evidence indicates that the level of oxidative stress is increased both systemically and in the myocardium of patients with heart failure. Oxidative stress in the heart may be due to reduced antioxidant capacity and/or the increased production of ROS, which may arise secondary to mechanical strain of the myocardium, neurohormonal stimulation (angiotensin II, alpha-adrenergic agonists, endothelin-1 [ET-1]) and/or inflammatory cytokines (tumor necrosis factor [TNF], interleukin [IL]-1. Excessive mitochondria-derived ROS in cardiac myocytes have been demonstrated in experimental models of heart failure and may contribute to contractile dysfunction in advanced heart failure. Increased xanthine oxidase expression and activity have been reported in canine rapid pacing–induced heart failure and patients with end-stage heart failure. Moreover, increased expression and activity of myocardial NADPH oxidases have recently been demonstrated in both experimental and human heart failure.5 In cultured cardiac myocytes ROS stimulate myocyte hypertrophy, reexpression of fetal gene programs, and apoptosis. ROS also can modulate fibroblast proliferation and collagen synthesis, and can trigger increased matrix metalloproteinase (MMP) abundance and activation. ROS also can affect the peripheral vasculature in heart failure by decreasing the bioavailability of NO. These and other observations have led to the suggestion that strategies to reduce ROS may be of therapeutic value in patients with heart failure. The NIH-sponsored EXACT-HF (NCT00987415) trial is examining the role of allopurinol in NYHA class II to IV heart failure patients with serum uric acid levels of 9.5 mg/dL or greater (a marker of oxidative stress). The importance of aldosterone, independent of angiotensin II, has been demonstrated by clinical trials (see Chapter 25) showing that low-dose spironolactone increased the survival of patients with NYHA class II to IV systolic heart failure, as well as improved survival after myocardial infarction, independent of changes in volume or electrolyte status.6 One of the signatures of advancing heart failure is increased salt and water retention by the kidneys. Traditional theories have ascribed this increase to either “forward” failure, which attributes sodium retention to inadequate renal perfusion as a consequence of impaired cardiac output, or “backward” failure, which emphasizes the importance of increased venous pressure in favoring transudation of salt and water from the intravascular to the extracellular compartment. These mechanisms have largely been supplanted by the concept of decreased effective arterial blood volume, which postulates that despite blood volume expansion in heart failure, inadequate cardiac output sensed by baroreceptors in the vascular tree leads to a series of compensatory neurohormonal adapations that resemble the homeostatic response to acute blood loss.7 As illustrated in Figure 22-6, a falling cardiac output and/or redistribution of the circulating blood volume is sensed by baroreceptors in the left ventricle, the aortic arch, the carotid sinus and the renal afferent arterioles. The loss of inhibitory input from arterial or cardiopulmonary baroreceptor reflexes leads to sustained activation of the sympathetic nervous and the renin-angiotensin systems. The ongoing XR-1 trial (NCT01484288) is using an implantable barostimulation device that activates the carotid baroreceptors to decrease sympathetic activation in patients with symptomatic heart failure, to determine whether this will restore the sympathovagal imbalance. There is little evidence to suggest that a primary renal abnormality is responsible for excessive sodium retention in heart failure. Rather, volume overload in heart failure probably is secondary to a functional derangement of renal physiology in response to several factors that have the potential to cause increased sodium reabsorption, including activation of the sympathetic nervous system, activation of RAS, reduced renal perfusion pressures, and blunting of renal responsiveness to natriuretic peptides. Increased renal sympathetic nerve–mediated vasconstriction leads to decreased renal blood flow, as well as increased renal tubular sodium and water reabsorption throughout the nephron. Renal sympathathetic stimulation also can lead to the nonosmotic release of arginine vasopressin (AVP) from the posterior pituitary, which reduces the excretion of free water and contributes to worsening peripheral vasoconstriction, as well as increased ET production.7 Increased renal sympathetic activity leads to increased renin production by the kidneys, with a resultant sustained activation of RAS, despite an expanded extracellular volume (see Fig. 22-3). Angiotensin II facilitates retention of sodium and water by multiple renal mechanisms, including a direct proximal tubular effect, as well as through activation of aldosterone, which leads to increased sodium resorption in the distal tubule. Angiotensin II also stimulates the thirst center of the brain and provokes the release of AVP and aldosterone, which both can lead to further dysregulation of salt and water homeostasis. A number of counterregulatory neurohormonal systems become activated in heart failure in order to offset the deleterious effects of the vasoconstricting neurohormones (Table e22-1 Natriuretic Peptides. The natriuretic peptide system consists of five structurally similar peptides collectively termed ANP, urodilantin (an isoform of ANP), BNP, C-type natriuretic peptide (CNP), and dendroaspis natriuretic peptide (DNP)9 (Fig. 22-7A). ANP, a 28-amino-acid peptide hormone, is produced principally in the cardiac atria, whereas BNP, a 32-amino-acid peptide originally isolated from porcine brain, was later identified as a hormone that was primarily produced in the cardiac ventricles.9 Both ANP and BNP are secreted in response to increasing cardiac wall tension; however, other factors such as neurohormones (e.g., angiototensin II, ET-1) or physiologic factors (e.g., age, sex, renal function) may also play a role in their regulation. The biosynthesis, secretion and clearance of BNP differs from ANP, suggesting that these two natriuretic peptides have discrete physiologic and pathophysiologic roles. Whereas ANP is secreted in short bursts in response to acute changes in atrial pressure, the activation of BNP is regulated transcriptionally in response to chronic increases in atrial/ventricular pressure. ANP and BNP initially are synthesized as prohormones that are subsequently proteolytically cleaved, respectively, by corin and furin, to yield large, biologically inactive N-terminal fragments (NT-ANP and NT-BNP) and smaller, biologically active peptides (ANP and BNP). ANP has a relatively short half-life of approximately 3 minutes, whereas BNP has a plasma half-life of approximately 20 minutes. C-type natriuretic peptide (CNP), which is located primarily in the vasculature, also is released as a prohormone that is cleaved into biologically inactive form (NT-CNP) and a 22-amino-acid biologically active form (i.e., CNP). Figure 22-7B illustrates the signaling pathway of the natriuretic peptide system. The natriuretic peptides stimulate the production of the intracellular second messenger cyclic guanosine monophosphate (cGMP), via binding to the natriuretic peptide A receptor (NPR-A), which preferentially binds ANP and BNP, and the natriuretic peptide B receptor (NPR-B), which preferentially binds CNP. Both NPR-A and NPR-B receptor are coupled to particulate guanylate cyclase. Activation of NPR-A and NPR-B results in natriuresis, vasorelaxation, inhibition of renin and aldosterone, inhibition of fibrosis, and increased lusitropy. The natriuretic peptide C receptor (NPR-C) is not linked to cGMP and serves as a clearance receptor for the natriuretic peptides. Natriuretic peptides are degraded by neutral endopeptidase (NEP) 24.11 (neprilysin), which is widely expressed in multiple tissues, where it often is colocalized with ACE. Inhibition of NEP may further potentiate the renal actions of ANP and BNP. The experience with omapatrilat, which inhibited both neutral endopeptidase and ACE, showed that omipatrilat was no more effective than ACE inhibitor alone in patients with heart failure.10 However, the use of a combined AT1 receptor antagonist and a neprilysin inhibitor (LCZ696) was shown to have a favorable impact on LV structure in patients with heart failure and a preserved ejection fraction (see Chapter 27). The biologic importance of the natriuretic peptides in renal sodium handling has been demonstrated in multiple studies using natriuretic peptide receptor antagonists, as well as overexpression of ANP or BNP. In experimental heart failure models, either acute blockade of the natriuretic A and B receptors or chronic genetic disruption of the natriuretic peptide A receptor blunts the renal natriuretic response to acute volume expansion, demonstrating the renal protective action of natriuretic peptide activation. The infusion of a recombinant human ANP and BNP exerts beneficial hemodynamic effects that are characterized by decreases in arterial and venous pressures, an increase in cardiac output, and suppression of neurohormonal activation in humans, resulting in their clinical development as therapeutic agents for human heart failure (see Chapter 24). In addition to their important biologic role, the natriuretic peptides have provided important diagnostic and prognostic information in heart failure (see Chapter 23). In patients with heart failure, the complex interactions between the autonomic nervous system and local autoregulatory mechanisms tend to preserve circulation to the brain and heart while decreasing blood flow to the skin, skeletal muscles, splanchnic organs, and kidneys. This intense visceral vasoconstriction during exercise helps to divert the limited cardiac output to exercising muscle but contributes to hypoperfusion of the gut and kidneys. The most powerful stimulus for peripheral vasoconstriction is sympathetic activation, which releases the potent vasoconstrictor NE. Other vasoconstrictors that contribute to maintaining circulatory homeostasis include angiotensin II, ET, neuropeptide Y, urotensin II, thromboxane A2, and AVP (see Table e22-1). The increased sympathetic adrenergic stimulation of the peripheral arteries and the increased concentrations of circulating vasoconstrictors contribute to the arteriolar vasoconstriction and to the maintenance of arterial pressure, while the sympathetic stimulation of the veins contributes to an increase in venous tone, which helps to maintain venous return and ventricular filling and to support cardiac performance by Starling’s law of the heart (Chapter 21). As noted previously, the vasoconstricting neurohormones activate counterregulatory vasodilator responses, including release of natriuretic peptides, NO, bradykinin, adrenomedullin, apelin, and vasodilating prostaglandins PGI2 and PGE2 (see Table e22-1). Under normal circumstances, the continuous release of NO (endothelium-derived relaxing factor) from the endothelium counteracts the vasoconstricting factors and allows for appropriate vasodilatory responses during exercise. As heart failure advances, however, the endothelial cell–mediated vasodilatory responsiveness is lost, which contributes to the excessive peripheral arterial vasoconstriction that is emblematic of advanced heart failure. Of interest, the vasodilator response can be restored by the administration of l-arginine, a precursor of endothelium-derived NO (NO). The free radical gas NO is produced by three isoforms of NO synthase (NOS). All three isoforms are present in the heart, including NOS1 (neuronal NOS [nNOS]), NOS2 (inducible NOS [iNOS]) and NOS3 (so-called endothelial-constitutive NOS [eNOS]). NOS1 has been detected in cardiac conduction tissue, in intracardiac neurons, and in the SR of cardiac myocytes; NOS2 is an inducible isoform that is not normally expressed in the myocardium but is synthesized de novo in virtually all cells in the heart in response to inflammatory cytokines, whereas NOS3 is expressed in coronary endothelium and endocardium and in the sarcolemma and T-tubule membranes of cardiac myocytes. NOS1 and NOS3 can be activated by calcium or calmodulin, whereas the induction of NOS2 is calcium-independent. NO activates soluble guanylate cyclase (see Fig. e22-2A The actions of NO on the myocardium are complex and include both short-term alterations in function and energetics and longer-term effects on structure. NO modulates the activity of several key calcium channels involved in excitation-contraction coupling as well as mitochondrial respiratory complexes. This type of regulation is accomplished by spatial localization of different NOS isoforms in distinct cellular microdomains involved in excitation-contraction coupling. Specifically, NOS1 localizes to the SR in proximity to the ryanodine receptor (RyR) and the SR Ca2+ ATPase (SERCA2a), and NOS3 is found in sarcolemmal caveolae compartmentalized with cell surface receptors and the L-type Ca2+ channel (Fig. e22-2B). NO also participates in mitochondrial respiration, the process that fuels excitation-contraction coupling. The different NOS isoforms also may participate in the process of cardiac remodeling. LV remodeling was ameliorated and survival improved after myocardial infarction in transgenic mice deficient in NOS2.14 By contrast, overexpression of NOS3 resulted in improved remodeling after myocardial infarction. These contrasting effects of NOS2 and NOS3 may reflect the differences in amount of NO produced, which is much higher with NOS2. Emerging evidence points to an imbalance between increasing free radical production and decreased NO generation in heart failure, which has been termed the nitroso-redox imbalance.15 NOS uncoupling secondary to a deficiency of tetrahydrobiopterin may further contribute to the nitroso-redox imbalance.14 The nitroso-redox imbalance probably contributes to disease progression in heart failure secondary to increased oxidative stress, as well as loss of the peripheral vasodilatory effects of NO. Bradykinin. Kinins are vasodilators that are released from inactive protein precursors (kininogens) through the action of proteolytic enzymes termed kallikreins. The biologic actions of the kinins are mediated by binding to B1 and B2 receptors. Most cardiovascular actions are initiated by the B2 receptor, which is distributed widely in tissues, where it binds bradykinin and kallidin. The B1 receptor binds the metabolites of bradykinin and kallidin. Stimulation of the B2 receptor leads to vasodilation, which is mediated by the activation of NOS3, phospholipase A2, and adenylyl cyclase. Studies suggest that bradykinin plays an important role in the regulation of vascular tone in heart failure.16 The breakdown of bradykinin is catalyzed by ACE, so that this enzyme not only leads to the formation of a potent vasoconstrictor (angiotensin II) but also mediates the breakdown of a vasodilator (bradykinin). The augmentation of bradykinin levels likely contributes to the beneficial actions of ACE inhibitors (see Chapter 25). Adrenomedullin. Adrenomedullin is a 52-amino-acid vasodilatory peptide that originally was discovered in human pheochromocytoma tissue. Subsequently, high levels of adrenomedullin immunoreactivity were detected in cardiac atrium and adrenal and pituitary glands, with lower levels detected in the ventricle, kidney, and vasculature.17 Adrenomedullin binds to a number of G protein–coupled receptors, including the calcitonin receptor-like receptor and one specific for the adrenomedullin peptide. Adrenomedullin receptors are present in multiple tissue beds, as well as both endothelial and vascular smooth muscle cells. Circulating concentrations of adrenomedullin are elevated in cardiovascular disease and heart failure in proportion to the severity of cardiac and hemodynamic impairment. Increasing evidence suggests that adrenomedullin may play a compensatory role in heart failure by offsetting the deleterious effects of excessive peripheral vasoconstriction. Plasma levels of adrenomedullin are elevated in chronic heart failure and are increased proportionally to disease severity. Immunoassays that detect the prohormone form of adrenomedullin have been shown to predict heart failure–realted death in the BAC (Biomarkers in Acute Heart Failure) trial.18 Apelin. Apelin is a vasoactive peptide that is an endogenous ligand for the G protein–coupled receptor APJ. In the cardiovascular system, apelin elicits endothelium-dependent, NO-mediated vasorelaxation and reduces arterial blood pressure. In addition, apelin demonstrates potent and inotropic activity without stimulating concomitant cardiac myocyte hypertrophy. Apelin also produces diuresis by inhibition of arginine vasopressin activity. In experimental animals, apelin concentrations are significantly lower in failing hearts and are increased after treatement with an angiotensin receptor blocking agent. Furthermore, apelin levels are significantly reduced in patients with heart failure when compared with control subjects and are significantly increased after cardiac resynchronization. The cognate receptor for apelin, the APJ receptor, is bifunctional G protein receptor that conveys cytoprotective signals after endogenous ligand stimulatio, and also acts a mechanosensor to delimit cardiac hypertrophy following hemodynamic pressure overload.19 Adipokines. Although adipose tissue was once considered as a simple storage depot for fat, adipose tissue is now known to synthesize and secrete a family of proteins collectively referred to as adipokines (see Fig. e22-3 Adiponectin is a 224-amino-acid polypeptide that modulates a number of metabolic processes, including glucose regulation and fatty acid oxidation. Although adiponectin initially was thought to be exclusively produced by adipose tissue, recent studies have demonstrated adiponectin expression in the heart. Studies in adiponectin-deficient mice demonstrated progressive cardiac remodeling after hemodynamic pressure overloading, whereas administration of adiponectin diminished the infarct size, apoptosis, and TNF production after myocardial ischemia-reperfusion in both wild-type and adiponectin-deficient mice. Of interest, many studies have correlated decreased adiponectin levels with the development of obesity-linked heart failure. Thus adiponectin has been proposed as a potential biomarker of heart failure and as a potential therapeutic target in its treatment.20 Inflammatory Mediators.
Pathophysiology of Heart Failure
Overview
Pathogenesis
Heart Failure as a Progressive Model
Neurohormonal Mechanisms
Activation of the Sympathetic Nervous System
 ). Although these disturbances in autonomic control initially were attributed to loss of the inhibitory input from arterial or cardiopulmonary baroreceptor reflexes, increasing evidence indicates that excitatory reflexes also may participate in the autonomic imbalance that occurs in heart failure.2 Under normal conditions, inhibitory inputs from “high-pressure” carotid sinus and aortic arch baroreceptors and the “low-pressure” cardiopulmonary mechanoreceptors are the principal inhibitors of sympathetic outflow, whereas discharge from the nonbaroreflex peripheral chemoreceptors and that from muscle metaboreceptors are the major excitatory inputs to sympathetic outflow. The vagal limb of the baroreceptor heart rate reflex also is responsive to arterial baroreceptor afferent inhibitory input. Healthy persons display low sympathetic discharge at rest and have a high heart rate variability. In patients with heart failure, however, inhibitory input from baroreceptors and mechanoreceptors decreases and excitatory input increases, with the net result of a generalized increase in sympathetic nerve traffic and blunted parasympathetic nerve traffic, leading to loss of heart rate variability and increased peripheral vascular resistance.2
). Although these disturbances in autonomic control initially were attributed to loss of the inhibitory input from arterial or cardiopulmonary baroreceptor reflexes, increasing evidence indicates that excitatory reflexes also may participate in the autonomic imbalance that occurs in heart failure.2 Under normal conditions, inhibitory inputs from “high-pressure” carotid sinus and aortic arch baroreceptors and the “low-pressure” cardiopulmonary mechanoreceptors are the principal inhibitors of sympathetic outflow, whereas discharge from the nonbaroreflex peripheral chemoreceptors and that from muscle metaboreceptors are the major excitatory inputs to sympathetic outflow. The vagal limb of the baroreceptor heart rate reflex also is responsive to arterial baroreceptor afferent inhibitory input. Healthy persons display low sympathetic discharge at rest and have a high heart rate variability. In patients with heart failure, however, inhibitory input from baroreceptors and mechanoreceptors decreases and excitatory input increases, with the net result of a generalized increase in sympathetic nerve traffic and blunted parasympathetic nerve traffic, leading to loss of heart rate variability and increased peripheral vascular resistance.2
Activation of the Renin-Angiotensin System
Neurohormonal Alterations of Renal Function
 ). Metabolites of vasodilatory prostaglandins, including prostaglandin E2 (PGE2) and prostacyclin (PGI2), are elevated in patients with heart failure. In addition to being a vasodilator, PGE2 enhances renal sodium excretion and modulates the antidiuretic action of AVP. One class of the most important counterregulatory neurohormonal systems that become activated in heart failure are the natriuretic peptides, including atrial natriuretic peptide (ANP) and brain type natriuretic peptide (BNP). Under physiologic conditions, ANP and BNP function as natriuretic hormones that are released in response to increases to atrial and or myocardial stretch, often secondary to excessive sodium intake. Once released, these cardiac peptides act on the kidney and peripheral circulation to unload the heart, through increased excretion of sodium and water, while inhibiting the release of renin and aldosterone. In the setting of RAS activation, the release of ANP and BNP may serve as an important counterregulatory mechanism that maintains sodium and water homeostasis. However, for reasons that are not entirely clear, the renal effects of the natriuretic peptides appear to become blunted with advancing heart failure, leaving the effects of RAS unopposed.8 Potential reasons for this blunting include low renal perfusion pressure, relative deficiency or altered molecular forms of the natriuretic peptides, and decreased levels of natriuretic peptide receptors.
). Metabolites of vasodilatory prostaglandins, including prostaglandin E2 (PGE2) and prostacyclin (PGI2), are elevated in patients with heart failure. In addition to being a vasodilator, PGE2 enhances renal sodium excretion and modulates the antidiuretic action of AVP. One class of the most important counterregulatory neurohormonal systems that become activated in heart failure are the natriuretic peptides, including atrial natriuretic peptide (ANP) and brain type natriuretic peptide (BNP). Under physiologic conditions, ANP and BNP function as natriuretic hormones that are released in response to increases to atrial and or myocardial stretch, often secondary to excessive sodium intake. Once released, these cardiac peptides act on the kidney and peripheral circulation to unload the heart, through increased excretion of sodium and water, while inhibiting the release of renin and aldosterone. In the setting of RAS activation, the release of ANP and BNP may serve as an important counterregulatory mechanism that maintains sodium and water homeostasis. However, for reasons that are not entirely clear, the renal effects of the natriuretic peptides appear to become blunted with advancing heart failure, leaving the effects of RAS unopposed.8 Potential reasons for this blunting include low renal perfusion pressure, relative deficiency or altered molecular forms of the natriuretic peptides, and decreased levels of natriuretic peptide receptors.
Neurohormonal Alterations in the Peripheral Vasculature
Nitric Oxide
 ). Under normal circumstances, the continuous release of NO (endothelium-derived relaxing factor) from the endothelium counteracts the vasoconstricting factors and allows for appropriate vasodilatory responses during exercise. This activation leads to the production of cGMP, which in turn activates protein kinase G and cascade of different signaling events. In normal subjects, NO released by endothelial cells mediates vasodilation in the peripheral vasculature through cGMP mediated relaxation of vascular smooth muscle. In patients with heart failure, endothelium-dependent NO-mediated dilation of the peripheral vasculature is blunted, which has been attributed to decreased NOS3 expression and activity.
). Under normal circumstances, the continuous release of NO (endothelium-derived relaxing factor) from the endothelium counteracts the vasoconstricting factors and allows for appropriate vasodilatory responses during exercise. This activation leads to the production of cGMP, which in turn activates protein kinase G and cascade of different signaling events. In normal subjects, NO released by endothelial cells mediates vasodilation in the peripheral vasculature through cGMP mediated relaxation of vascular smooth muscle. In patients with heart failure, endothelium-dependent NO-mediated dilation of the peripheral vasculature is blunted, which has been attributed to decreased NOS3 expression and activity.
 ). Adipokines include adiponectin, TNF, plasminogen activator inhibitor type 1 (PAI-1), transforming growth factor-β, and resistin. Leptin is a 16-kDa protein hormone that plays a key role in regulating energy intake and energy expenditure. Leptin, the product of the ob gene, is predominantly synthesized and secreted by adipocytes, although the heart is also a site of leptin synthesis. The initial role of leptin was thought to be decreasing appetite through hypothalamic stimulation and hence regulating food intake. However, elevated circulating levels of leptin, which act via a family of receptor (ob.R) isoforms, appear to play an important role in hypertension, hypertrophy, and heart failure.20 Leptin may affect myocardial function through direct peripheral effects or through secondary central nervous system–mediated responses. Lack of leptin and/or leptin resistance may lead to an accumulation of lipids in nonadipose peripheral tissues, resulting in a variety of “lipotoxic” effects, including cardiac myocyte apoptosis. Several studies suggest that leptin directly induces hypertrophy in both human and rodent cardiac myocytes.20
). Adipokines include adiponectin, TNF, plasminogen activator inhibitor type 1 (PAI-1), transforming growth factor-β, and resistin. Leptin is a 16-kDa protein hormone that plays a key role in regulating energy intake and energy expenditure. Leptin, the product of the ob gene, is predominantly synthesized and secreted by adipocytes, although the heart is also a site of leptin synthesis. The initial role of leptin was thought to be decreasing appetite through hypothalamic stimulation and hence regulating food intake. However, elevated circulating levels of leptin, which act via a family of receptor (ob.R) isoforms, appear to play an important role in hypertension, hypertrophy, and heart failure.20 Leptin may affect myocardial function through direct peripheral effects or through secondary central nervous system–mediated responses. Lack of leptin and/or leptin resistance may lead to an accumulation of lipids in nonadipose peripheral tissues, resulting in a variety of “lipotoxic” effects, including cardiac myocyte apoptosis. Several studies suggest that leptin directly induces hypertrophy in both human and rodent cardiac myocytes.20
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Pathophysiology of Heart Failure
22