Jacques Genest, Peter Libby
Lipoprotein Disorders and Cardiovascular Disease
Lipids constitute approximately 70% (by mass) of the dry weight of plasma. Amino acids (proteins), nucleic acids, and carbohydrate make up the remainder. Approximately half of circulating lipids are sterols, with the other major components being glycerophospholipids (phospholipids) and glycerolipids (triglycerides), which circulate in lipoproteins.1 Thus vascular endothelial cells are continuously exposed to circulating lipoproteins, and the interaction between lipoproteins and cells of the arterial wall have major importance in the pathogenesis of human atherosclerosis (see also Chapter 41).
Accumulating data affirm the basic tenets of the “lipid hypothesis.” Observational data show a strong and consistent association across populations between elevated plasma (or serum) levels of cholesterol and low-density lipoprotein cholesterol (LDL-C) and cardiovascular disease, especially coronary artery disease (CAD). Chapter 42 presents the epidemiologic observational data on plasma lipids and lipoprotein lipids being a key component of cardiovascular risk factors. Experimental animal data show that the development of atherosclerosis requires cholesterol. Mendelian randomization analyses provide strong support of causality for genes related to LDL-C levels. Reduction of LDL-C levels reduces the risk for CAD, and the effect size is associated with the magnitude of the reduction in LDL-C.2 Thus low-density lipoprotein (LDL) meets the modified Koch postulates as a causal risk factor for atherosclerotic cardiovascular disease (CVD).
Chapter 41 discusses the biologic basis and pathophysiology of atherosclerosis. This chapter deals with the fundamentals of lipid metabolism, therapeutic approaches to the treatment of lipid disorders, and the evidence base regarding their clinical use.
The terms dyslipidemia and dyslipoproteinemia reflect disorders of the lipid and lipoprotein transport pathways associated with arterial disease more appropriately than does the term hyperlipidemia, which has long been used in clinical practice. Dyslipidemia encompasses disorders often encountered in clinical practice, such as a low high-density lipoprotein cholesterol (HDL-C) level and elevated triglyceride level but an average total plasma cholesterol level. Dyslipidemia also includes elevated lipoprotein(a) (Lp[a]) and uncommon genetic or acquired disorders of lipoprotein metabolism. Certain rare lipoprotein disorders can cause overt clinical manifestations, but most common dyslipoproteinemias themselves seldom cause symptoms or produce clinical signs that are evident on physical examination. Rather, they require laboratory tests for detection. Proper recognition and management of dyslipoproteinemias can reduce cardiovascular and total mortality rates. The fundamentals of lipidology presented here have importance for the daily practice of cardiovascular medicine.
Lipoprotein Transport System
Biochemistry of Lipids
Lipids are insoluble in water and soluble in organic solvents. Biologic lipids usually refer to a broad grouping of naturally occurring molecules that include fatty acids, waxes, eicosanoids, monoglycerides, diglycerides, triglycerides, phospholipids, sphingolipids, sterols, terpenes, prenols, and fat-soluble vitamins (A, D, E, and K), in contrast to the other major groupings of biologic molecules, namely, nucleic acids, proteins, amino acids, and carbohydrates. The major biologic functions of lipids include critical contributions to biologic membranes, energy storage, and the backbones or modifiers of many signaling molecules. Certain lipids, especially fatty acid, readily undergo oxidation and can generate substances highly toxic to cells. Fatty acids can be degraded in the mitochondrion by beta-oxidation, whereas the sterol nucleus resists enzymatic degradation. Cholesterol must therefore be modified into bile acids or hormones or be shed with the skin to be eliminated.
The lipid transport system has evolved in animals over eons of evolution to carry hydrophobic molecules (fat) from sites of origin (the intestinal system) to sites of utilization (muscles, rapidly dividing tissues, and hormone-producing tissues) through the aqueous (water) environment of plasma. The proteins that mediate this process, termed apolipoproteins, show conservation throughout evolution in organisms with a circulatory system. Most apolipoproteins are derived from an ancestral gene and contain both hydrophilic and hydrophobic domains. This amphipathic structure enables these proteins to bridge the interface between the aqueous environment of plasma and the phospholipid constituents of lipoprotein. The major types of lipids that circulate in plasma include cholesterol and cholesteryl esters, glycerophospholipids, sphingolipids, and glycerolipids (triglycerides) (Fig. 45-1). The LIPID MAPS (Lipid Metabolites and Pathways Strategy) consortium has provided standardized nomenclature for lipids.3

Cholesterol is an essential component of mammalian cell membranes and the membranes of subcellular organelles (endoplasmic reticulum, Golgi, mitochondria, lysosomes, nucleus, endosomes, peroxisomes) and furnishes the substrate for steroid hormones and bile acids. Many cell functions depend critically on membrane cholesterol, and cells tightly regulate their cholesterol content. Most of the cholesterol in plasma circulates in the form of cholesteryl esters in the core of lipoprotein particles. The enzyme lecithin-cholesterol acyltransferase (LCAT) forms cholesteryl esters in the blood compartment by transferring a fatty acyl chain from phosphatidylcholine to cholesterol.
Glycerolipids (triglycerides) consist of a three-carbon glycerol backbone covalently linked to three fatty acid chains (designated R1, R2, and R3). The fatty acid composition varies in terms of chain length and the presence of double bonds (degree of saturation). Triglyceride molecules are nonpolar and hydrophobic; they are transported in the core of the lipoprotein. Hydrolysis of triglycerides by lipases generates the free fatty acids (FFAs) used for energy.
Glycerophospholipids are constituents of all cellular membranes and consist of a glycerol molecule linked to two fatty acids (designated R1 and R2; see Fig. 45-1). The fatty acyl residue in position sn-1 is usually a saturated fatty acid residue. Fatty acids differ in length and in the presence of a single (monounsaturated) or multiple (polyunsaturated) double bonds. The third carbon (sn-3) of the glycerol moiety carries a phosphate group to which one of four molecules is linked: choline (phosphatidylcholine, also called lecithin), ethanolamine (phosphatidylethanolamine), serine (phosphatidylserine), or inositol (phosphatidylinositol). More complex phospholipids include phosphatidylglycerol (cardiolipin is formed by the fusion of two phosphatidylglycerol molecules—antibodies against cardiolipin are often found in systemic lupus), and plasmalogens, an important constituent of eukaryotic membranes. Another phospholipid, sphingomyelin, has special functions in the plasma membrane in the formation of membrane microdomains such as rafts and caveolae. The structure of sphingomyelin resembles that of phosphatidylcholine. The backbone of sphingolipids uses the amino acid serine rather than glycerol. Phospholipids are polar molecules, more soluble than triglycerides or cholesterol or its esters. Phospholipids participate in signal transduction pathways: hydrolysis by membrane-associated phospholipases generates second messengers, including diacylglycerols, lysophospholipids, phosphatidic acids, and FFAs such as arachidonate, that regulate many cell functions. The phosphorylation of phosphatidylinositol contributes critically to membrane and cell organelle signaling and transport.
Lipoproteins, Apolipoproteins, Receptors, and Processing Enzymes
Lipoproteins are complex macromolecular structures composed of an envelope of phospholipids and free cholesterol and a core of cholesteryl esters and triglycerides. Apolipoproteins constitute the protein moiety of lipoproteins (Fig. 45-2). Lipoproteins vary in size, density in the aqueous environment of plasma, and lipid and apolipoprotein content (Fig. 45-3, Table 45-1). The classification of lipoproteins reflects their density in plasma (the density of plasma is 1.006 g/mL) as gauged by flotation in an ultracentrifuge. The triglyceride-rich lipoproteins (TRLs), which consist of chylomicrons, chylomicron remnants, and very low-density lipoprotein (VLDL), have a density of less than 1.006 g/mL. The rest (bottom fraction) of the ultracentrifuged plasma consists of LDL, high-density lipoprotein (HDL), and Lp(a).


TABLE 45-1
Plasma Lipoprotein Composition
| ORIGIN | DENSITY (g/mL) | SIZE (nm) | % PROTEIN | [CHOLESTEROL] IN PLASMA (mmol/L)* | [TRIGLYCERIDE] IN FASTING PLASMA (mmol/L)† | MAJOR APO | OTHER APO | |
| Chylomicrons‡ | Intestine | <0.95 | 100-1000 | 1-2 | 0.0 | 0 | B48 | A-I, C’s |
| Chylomicron remnants‡ | Chylomicron metabolism | 0.95-1.006 | 30-80 | 3-5 | 0.0 | 0.0 | B48, E | A-I, A-IV, C’s |
| VLDL | Liver | <1.006 | 40-50 | 10 | 0.1-0.4 | 0.2-1.2 | B100 | A-I, C’s |
| IDL | VLDL | 1.006-1.019 | 25-30 | 18 | 0.1-0.3 | 0.1-0.3 | B100, E | |
| LDL | IDL | 1.019-1.063 | 20-25 | 25 | 1.5-3.5 | 0.2-0.4 | B100 | |
| HDL | Liver, intestine | 1.063-1.210 | 6-10 | 40-55 | 0.9-1.6 | 0.1-0.2 | A-I, A-II | A-IV |
| Lp(a) | Liver | 1.051-1.082 | 25 | 30-50 | B100, (a) |

* In mmol/L; for mg/dL, multiply by 38.67.
† In mmol/L; for mg/dL, multiply by 88.5.
‡ In the fasted state, serum (or plasma) should not contain chylomicrons or their remnants.
APO = apolipoprotein; HDL = high-density lipoprotein; IDL = intermediate-density lipoprotein; VLDL = very low-density lipoprotein.
Apolipoproteins have four major roles: (1) assembly and secretion of the lipoprotein (apo A-I, B100, and B48), (2) structural integrity of the lipoprotein (apo B, E, A-I, and A-II), (3) coactivators or inhibitors of enzymes (apo A-I, A-V, C-I, C-II, and C-III), and (4) binding or docking to specific receptors and proteins for cellular uptake of the entire particle or selective uptake of a lipid component (apo A-I, B100, and E) (Table 45-2). The role of several apolipoproteins (A-IV, A-V, D, H, J, L, and M) remains incompletely understood.

Many proteins regulate the synthesis, secretion, and metabolic fate of lipoproteins; their characterization has provided insight into molecular cellular physiology and targets for drug development (Table 45-3). Discovery of the LDL receptor (LDL-R) represented a landmark in understanding cholesterol metabolism and receptor-mediated endocytosis.4 LDL-R regulates the entry of cholesterol into cells, and tight control mechanisms alter its expression on the cell surface, depending on need. LDL-R belongs to a superfamily of membrane receptors that includes LDL-R, VLDL-R, LDL-R–mediated peptide type 1 (LRP1; apo E receptor), LRP1B, LRP4 (MGEF7), LRP5 and LRP6 (involved in the process of bone formation), LRP8 (apo E receptor-2), and LRP9.5 LRP1, which mediates the uptake of chylomicron remnants and VLDL, preferentially recognizes apo E. LPR1 also interacts with hepatic lipase. The interaction between hepatocytes and the various lipoproteins containing apo E is complex and involves cell surface proteoglycans that provide scaffolding for lipolytic enzymes (lipoprotein lipase [LPL] and hepatic lipase) involved in recognition of remnant lipoproteins. Macrophages express receptors that bind modified (especially oxidized) lipoproteins. These scavenger lipoprotein receptors mediate the uptake of oxidatively modified LDL into macrophages. In contrast to the exquisitely regulated LDL-R, high cellular cholesterol content does not suppress scavenger receptors, thereby enabling intimal macrophages to accumulate abundant cholesterol, become foam cells, and form fatty streaks. Sterol accumulation in the endoplasmic reticulum may lead to cell apoptosis via the unfolded protein response.6 Endothelial cells can also take up modified lipoproteins through a specific receptor, such as the oxidized LDL-R LOX-1.

At least three physiologically relevant receptors bind HDL particles: the scavenger receptor class B (SR-B1) and the adenosine triphosphate (ATP)-binding cassette transporters A1 (ABCA1) and G1 (ABCG1). SR-B1 is a receptor for HDL (also for LDL and VLDL, but with less affinity). SR-B1 mediates the selective uptake of HDL cholesteryl esters in steroidogenic tissues, hepatocytes, and endothelium. ABCA1 mediates cellular phospholipid (and possibly cholesterol) efflux and is necessary and essential for HDL biogenesis. The ABCG1 transporter transfers cellular cholesterol to spherical HDL particles.
Lipoprotein Metabolism and Transport
The lipoprotein transport system has two major roles: efficient transport of triglycerides from the intestine and liver to sites of utilization (fat tissue or muscle), and transport of cholesterol to peripheral tissues for membrane synthesis and steroid hormone production or to the liver for bile acid synthesis (Fig. 45-4).
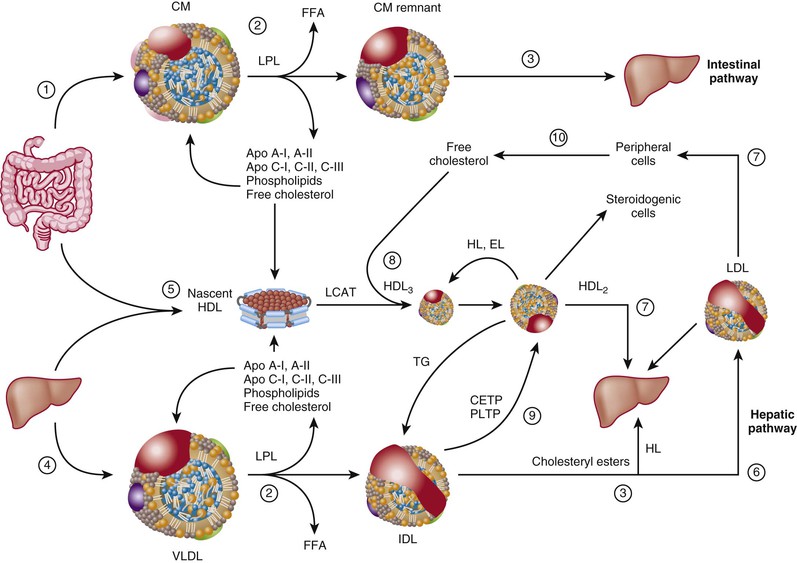
Intestinal Pathway (Chylomicrons to Chylomicron Remnants)
Life requires fats. The human body derives essential fatty acids (linoleic acid, from which arachidonic acid is derived, and linolenic acid, which leads to the formation of eicosapentaenoic acid) that it cannot make from the diet. Fat typically furnishes 20% to 40% of daily calories. Triglycerides account for the major portion of ingested fats. For an individual consuming 2000 kcal/day, with 30% in the form of fat, this represents approximately 66 g of triglycerides and 250 mg (0.250 g) of cholesterol per day. The intestine has very efficient fat absorption mechanisms, probably evolved to maximize provision of the organism with nutrients under circumstances of limited or irregular availability of food.
On ingestion, lingual and pancreatic lipases hydrolyze triglycerides into FFAs and monoglycerides or diglycerides. Emulsification by bile salts leads to the formation of intestinal micelles. Micelles resemble lipoproteins in that they consist of phospholipids, free cholesterol, bile acids, diglycerides and monoglycerides, FFAs, and glycerol. The mechanism of micelle uptake by intestinal brush border cells still engenders debate. The Niemann-Pick C1-like 1 (NPC1L1) protein is part of an intestinal cholesterol transporter complex and the target for the selective cholesterol absorption inhibitor ezetimibe (see later).7 After uptake into intestinal cells, fatty acids undergo re-esterification to form triglycerides and packaging into chylomicrons inside the intestinal cell and enter the portal circulation (Fig. 45-4, path 1). Chylomicrons contain apo B48, the amino-terminal component of apo B100. In the intestine, the apo B gene is modified during transcription into mRNA by substitution of a uracil for a cytosine via an apo B48–editing enzyme complex (ApoBec). This mechanism involves a cytosine deaminase and leads to a termination codon at residue 2153 and a truncated form of apo B. Only intestinal cells express ApoBec. Apo B48 does not bind to LDL-R. Intestinal cells absorb plant sterols (sitosterol, campesterol), sort these compounds into a separate cellular compartment, and resecrete them into the intestinal lumen via the ABCG5/8 heterodimeric transporter. Mutations of the ABCG5/8 genes cause the rare disorder sitosterolemia.
Chylomicrons rapidly enter the plasma compartment after meals. In capillaries of adipose tissue or muscle cells in the peripheral circulation, chylomicrons encounter LPL, an enzyme attached to heparan sulfate proteoglycans, and present on the luminal surface of endothelial cells (Fig. 45-4, path 2). LPL activity is modulated by apo C-II and apo A-V (activators) and by apo C-III (an inhibitor). LPL has broad specificity for triglycerides; it cleaves all fatty acyl residues attached to glycerol, and in the process generates three molecules of FFA for each molecule of glycerol. Muscle cells rapidly take up fatty acids. Fatty acids provide the energy substrate for muscle contraction by the generation of ATP during beta-oxidation of fatty acyl residues in mitochondria. Adipose cells can store triglycerides made from fatty acids for energy utilization, a process that requires insulin. Conversely, hormone-sensitive lipase is a triglyceride lipase that is activated by cyclic adenosine monophosphate (cAMP) in response to stress and releases fatty acids from adipose tissues. Fatty acids can also bind to fatty acid–binding proteins and albumin and travel to the liver, where they are repackaged in VLDL. Peripheral resistance to insulin can thus increase the delivery of FFAs to the liver with a consequent increase in VLDL secretion and increased apo B particles in plasma. As discussed later, this is one of the consequences of metabolic syndrome and type 2 diabetes. The remnant particles, derived from chylomicrons following LPL action, contain apo E and enter the liver for degradation and reutilization of their core constituents (Fig. 45-4, path 3).
Hepatic Pathway (Very Low-Density Lipoprotein to Intermediate-Density Lipoprotein)
Food is not always available, and dietary fat content varies. The body must ensure that triglyceride is readily available to meet energy demands. Hepatic secretion of VLDL particles serves this function (Fig. 45-4, path 4). VLDLs are TRLs smaller than chylomicrons (see Table 45-1 and Fig. 45-3). They contain apo B100 as their main lipoprotein. As opposed to apo B48, apo B100 contains a domain recognized by LDL-R (the apo B/E receptor). VLDL particles follow the same catabolic pathway through LPL as chylomicrons do (see Fig. 45-4, path 2). During hydrolysis of TRLs by LPL, an exchange of proteins and lipids takes place: VLDL particles (and chylomicrons) acquire apo C’s and apo E, in part from HDL particles. VLDLs also exchange triglycerides for cholesteryl esters from HDL (mediated by cholesteryl ester transfer protein [CETP]) (Fig. 45-4, path 9). Such bidirectional transfer of constituents between lipoproteins serves several purposes: acquisition of specific apolipoproteins by lipoproteins that will dictate their metabolic fate, transfer of phospholipids onto nascent HDL particles mediated by phospholipid transfer protein (PLTP) (during loss of the core triglycerides, the phospholipid envelope becomes redundant and sheds apo A-I to form new HDL particles), and transfer of cholesterol from HDL to VLDL remnants so that it can be metabolized in the liver. This exchange constitutes a major part of the “reverse cholesterol transport pathway.”
After hydrolysis of triglycerides partly depletes VLDL of triglycerides, VLDL particles have relatively more cholesterol, shed several apolipoproteins (especially the C apolipoproteins), and acquire apo E. The VLDL remnant lipoprotein, called intermediate-density lipoprotein (IDL), is taken up by the liver via its apo E moiety (see Fig. 45-4, path 3) or is further delipidated by hepatic lipase to form an LDL particle (Fig. 45-4, path 6). At least four receptors take up TRLs, TRL remnants, and apo B–containing lipoproteins: VLDL-R, the remnant receptor (apo ER2), LDL-R (also called the apo B/E receptor), and LRP1. Most hepatic receptors share the ability to recognize apo E, an engagement that mediates the uptake of several classes of lipoproteins, including VLDL and IDL.5 The interaction between apo E and its ligand is complex and involves the “docking” of TRLs on heparan sulfate proteoglycans before presentation of the ligand to its receptor.
Low-Density Lipoproteins
LDL particles contain predominantly cholesteryl esters packaged with the protein moiety apo B100. Normally, triglycerides constitute only 4% to 8% of the LDL mass (see Table 45-1). In the presence of elevated plasma triglyceride levels, LDL particles can become enriched in triglycerides and depleted in core cholesteryl esters. Variation in LDL particle size results from changes in core constituents, with an increase in triglycerides and a relative decrease in cholesteryl esters leading to smaller, denser LDL particles.
Humans are unusual among mammals in that they use LDL as a major cholesterol transporter. Nonhuman primates fed a cholesterol-enriched diet also carry cholesterol in LDL. In other mammals, such as rodents or rabbits, VLDL carries triglycerides, and HDL particles transport most of the cholesterol. Cells can either make cholesterol from acyl coenzyme A (CoA) through enzymatic reactions requiring at least 33 steps, or obtain it as cholesteryl esters from HDL and LDL particles. Cells internalize LDL via LDL-R (Fig. 45-5A). LDL particles contain one molecule of apo B. Although several domains of apo B are highly lipophilic and associated with phospholipids, a region surrounding residue 3500 binds with high affinity to LDL-R. LDL-R is localized in a region of the plasma membrane rich in the protein clathrin (Fig. 45-5A; also Fig. 45-4, path 7). Once bound to the receptor, clathrin polymerizes and forms an endosome that contains LDL bound to its receptor, a portion of the plasma membrane, and clathrin. This internalized particle then fuses with lysosomes whose hydrolytic enzymes (cholesteryl ester hydrolase, cathepsins) release free cholesterol and degrade apo B. LDL-R will detach itself from its ligand and recycle to the plasma membrane.


Cells tightly regulate their cholesterol content by (1) synthesis of cholesterol in the smooth endoplasmic reticulum (via the rate-limiting step hydroxymethylglutaryl-CoA [HMG-CoA] reductase), (2) receptor-mediated endocytosis of LDL (two mechanisms under the control of steroid-responsive element binding protein-2 [SREBP-2]), (3) efflux of cholesterol from the plasma membrane to cholesterol acceptor particles (predominantly apo A-I and HDL) via the ABCA1 and ABCG1 transporters, and (4) intracellular cholesterol esterification via the enzyme acetyl-CoA acetyltransferase (ACAT) (Fig. 45-5A-C). SREBP-2 coordinately regulates the first two pathways at the level of gene transcription. Cellular cholesterol binds to SCAP (SREPB cholesterol-activated protein), which is localized on the endoplasmic reticulum. Cholesterol inhibits the interaction of SCAP with SREPB. In the absence of cholesterol, SCAP mediates the cleavage of SREBP at two sites by specific proteases with the release of an amino (NH2) fragment of SREBP. The SREBP NH2 fragment migrates to the nucleus and increases the transcriptional activity of genes involved in cellular cholesterol and fatty acid homeostasis. Cleavage of SREBP depends on a proprotein convertase related to the subtilisin/kexin family of convertases. Another member of the convertase superfamily, proprotein convertase subtilisin/kexin 9 (PCSK9), regulates the internalization and cellular processing of LDL-R; gain-of-function mutations in this gene cause autosomal dominant hypercholesterolemia, whereas loss-of-function mutations increase LDL-R and lower LDL-C significantly.8 The ACAT pathway regulates the cholesterol content in membranes. Humans express two separate forms of ACAT. ACAT1 and ACAT2 are derived from different genes and mediate cholesterol esterification in cytoplasm and in the endoplasmic reticulum lumen for lipoprotein assembly and secretion.
Regulation of cholesterol efflux from cells depends in part on the ABCA1 pathway, controlled in turn by hydroxysterols (especially 24- and 27-OH cholesterol, which act as ligands for the liver-specific receptor [LXR] family of transcriptional regulatory factors). In conditions of cholesterol sufficiency, the cell can decrease its input of cholesterol by decreasing the de novo synthesis of cholesterol. The cell can also decrease the amount of cholesterol that enters the cell via the LDL-R, thereby increasing the amount stored as cholesteryl esters, and promote the removal of cholesterol by increasing its movement to the plasma membrane for efflux.
High-Density Lipoprotein and Reverse Cholesterol Transport
Epidemiologic studies have consistently shown an inverse relationship between plasma levels of HDL-C and the presence of CAD. HDL promotes reverse cholesterol transport and can prevent lipoprotein oxidation, exerts anti-inflammatory actions in vitro, and promotes cell proliferation and survival.9 In vitro, HDL has potent effects on vascular endothelial cells. It promotes the production of nitric oxide (NO) through several mechanisms and prevents the expression of vascular endothelial cell inflammatory mediators via modulation of nuclear factor kappa B (NF-κB). Mendelian randomization analyses have cast doubt on HDL’s causality as a protective cardiovascular risk factor. Mutations of the genes for ABCA1 (causing HDL deficiency) do not impart additional cardiovascular risk, and conversely, genetic polymorphisms of genes that increase HDL-C are not associated with protection from cardiovascular events.10
HDL has a complex and incompletely understood metabolism. The complexity arises because HDL particles acquire their components from several sources and these components undergo metabolism at different sites. In addition, steady-state levels of HDL in plasma may reflect the dynamic state of HDL-mediated cholesterol trafficking, in contrast to the situation with LDL. The intestine and liver synthesize apo A-I, the main protein of HDL. Approximately 80% of HDL originates from the liver and 20% from the intestine (Fig 45-4, path 5). Lipid-free apo A-I acquires phospholipids from cell membranes and from redundant phospholipids shed during the hydrolysis of TRLs. Lipid-free apo A-I binds to ABCA1 and promotes its phosphorylation via cAMP, which increases the net efflux of phospholipids and cholesterol onto apo A-I to form a nascent HDL particle (see Fig. 45-4, path 5). This particle contains apo A-I, phospholipids, and some free cholesterol (Fig. 45-5C).11 These nascent HDL particles will mediate further cellular efflux of cholesterol. Currently, standard laboratory tests do not measure these HDL precursors because they contain little or no cholesterol. On reaching a cell membrane, the nascent HDL particles capture membrane-associated cholesterol and promote the efflux of free cholesterol onto other HDL particles (Fig. 45-4, path 0). Conceptually, the formation of HDL particles appears to involve two steps. The first step involves binding of HDL apo A-I to ABCA1 and generation of a specific membrane microdomain that allows the subsequent lipidation of apo A-I.12 Efflux of cellular cholesterol from peripheral cells, such as macrophages, does not contribute importantly to overall HDL-C mass but may have an important effect on export of cholesterol from atheromas. Macrophages can transfer cholesterol to apo A-I and apo E, to nascent discoid or ellipsoid HDL particles via the ABCA1 transporter, or to more mature spherical HDL particles via the ABCG1 transporter (Fig 45-5D). The ABCG1 transporter does not promote cellular cholesterol efflux to lipid-free or lipid-poor apo A-I, but rather to mature HDL particles. HDL-mediated cellular cholesterol can be measured in plasma and is altered in many disease states, including diabetes and CAD.13 The plasma enzyme LCAT, an enzyme activated by apo A-I, then esterifies the free cholesterol (Fig. 45-4, path 8, and see Fig. 45-5C). LCAT transfers an acyl chain (a fatty acid) from the R2 position of a phospholipid to the 3′-OH residue of cholesterol, which results in the formation of a cholesteryl ester (see Fig. 45-1). In a process called selective uptake of cholesterol, HDL also provides cholesterol to steroid hormone–producing tissues and the liver through the scavenger receptor SR-B1 (see Fig. 45-5B).
Because of their hydrophobicity, cholesteryl esters move to the core of the lipoprotein, and the HDL particle now assumes a spherical configuration (a particle denoted HDL3). With further cholesterol esterification, the HDL particle increases in size to become the more buoyant HDL2. The cholesterol within HDL particles can be exchanged with TRLs via CETP, which mediates an equimolar exchange of cholesterol from HDL to TRL and movement of triglyceride from TRL onto HDL (see Fig. 45-4, path 9). Inhibition of CETP increases HDL-C in blood and has undergone exploration as a therapeutic target for prevention of CVD. PLTP mediates the transfer of phospholipids between TRL and HDL particles. Triglyceride-enriched HDLs are denoted HDL2b. Hepatic lipase can hydrolyze triglycerides and endothelial lipase can hydrolyze phospholipids within these particles and thereby convert them back to HDL3 particles.
Reverse cholesterol transport involves the uptake of cellular cholesterol from extrahepatic sources, such as lipid-laden macrophages, and its esterification by LCAT, transport by large HDL particles, and exchange for one triglyceride molecule by CETP. Hepatic receptors can now take up the cholesterol molecule originally on an HDL particle and residing in a TRL or LDL particle after this exchange. HDL particles therefore act as shuttles between tissue cholesterol, TRL, and the liver.
Reverse cholesterol transport by HDL constitutes a small but potentially important portion of the plasma HDL mass. Indeed, selective inactivation of macrophage ABCA1 does not change HDL-C levels in mice but increases atherosclerosis. The catabolism of HDL particles has engendered debate among lipoprotein researchers. The protein component of HDL particles is exchangeable with lipoproteins of other classes. The kidneys appear to be a route of elimination of apo A-I and other HDL apolipoproteins. The lipid components of HDL particles also follow a different metabolic route (Fig. 45-5B-D).
Lipoprotein Disorders
Definitions
Time and new knowledge have stimulated changes in the classification of lipoprotein disorders. The original classification of lipoprotein disorders by Fredrickson, Lees, and Levy (1967) was based on measurement of total plasma cholesterol and triglycerides and analyzed lipoprotein patterns after separation by electrophoresis. This classification recognized elevations of chylomicrons (type I), VLDL or pre-beta lipoproteins (type IV), beta lipoproteins (LDL) (type II), and both chylomicrons and VLDL (type V), as well as “broad beta” disease (or type III hyperlipoproteinemia). In addition, the combined elevation of pre-beta (VLDL) and beta (LDL) lipoproteins was recognized as type IIb hyperlipoproteinemia. Despite providing a useful conceptual framework, this classification has many drawbacks: it does not include HDL-C, and it does not differentiate severe monogenic lipoprotein disorders from the more common polygenic disorders. Subsequently, the World Health Organization, the European Atherosclerosis Society, and more recently, the National Cholesterol Education Program (NCEP) classified lipoprotein disorders on the basis of arbitrary cut points.
A practical approach describes the lipoprotein disorder by the absolute plasma levels of lipids (cholesterol and triglycerides) and lipoprotein cholesterol levels (LDL-C and HDL-C) and considers clinical manifestations of hyperlipoproteinemia in the context of biochemical characterization. For example, a young patient with eruptive xanthomas and a plasma triglyceride level of 11.3 mmol/L (1000 mg/dL) probably has familial hyperchylomicronemia as a result of LPL deficiency or other genetic defects. An obese, hypertensive middle-aged man with a cholesterol level of 6.4 mmol/L (245 mg/dL), a triglyceride level of 3.1 mmol/L (274 mg/dL), an HDL-C level of 0.8 mmol/L (31 mg/dL), and a calculated LDL-C level of 4.2 mmol/L (162 mg/dL) probably has metabolic syndrome, and this should trigger the clinician to seek other components of this cluster, including hypertension and hyperglycemia. Conversely, an obese middle-aged man with a plasma triglyceride level of 7 mmol/L (620 mg/dL) probably has mutations in several genes associated with plasma triglyceride levels.14
The clinical usefulness of apolipoprotein levels has stirred debate (see also Chapter 42). Taken as a single measurement, the apo B level provides information on the number of potentially atherogenic particles and can be used as a goal of lipid-lowering therapy. Similarly, LDL particle size correlates highly with plasma HDL-C and triglyceride levels, and most studies do not show it to be an independent cardiovascular risk factor. Small, dense LDL particles tend to track with features of metabolic syndrome, which usually involves dyslipoproteinemia with elevated plasma triglyceride and reduced HDL-C levels. The Emerging Risk Factors Collaboration studies have shown that measurement of non–HDL-C is equivalent to measurement of apo B in determination of cardiovascular risk. Indeed, measurement of non–HDL-C captures the cholesterol content in apo B–containing lipoproteins. Similarly, HDL-C tracks as well with CVD risk as apo A-I does.15 Such observations prompted a joint statement from the American Heart Association and the American College of Cardiology on the lack of incremental values of apolipoprotein measurement or lipoprotein particle size in predicting cardiovascular risk.16
Genetic Lipoprotein Disorders
Understanding of the genetics of lipoprotein metabolism has expanded rapidly. Classification of genetic lipoprotein disorders usually requires a biochemical phenotype in addition to a clinical phenotype. With the exception of familial hypercholesterolemia (FH), monogenic disorders tend to be infrequent or very rare. Disorders considered heritable on careful family study may be difficult to characterize unambiguously because of age, sex, penetrance, and gene-gene and environmental interactions. Most common lipoprotein disorders encountered clinically result from the interaction of increasing age, lack of physical exercise, weight gain, and a suboptimal diet with individual genetic makeup. Genetic lipoprotein disorders can either raise or lower levels of LDL, Lp(a), remnant lipoproteins, TRLs (chylomicrons and VLDL), or HDL (Table 45-4).
TABLE 45-4
Genetic Lipoprotein Disorders
| DISORDER | GENE | FIGURE 45-4 |
| LDL Particles | ||
| Autosomal dominant hypercholesterolemia (ADH) | ||
| Familial hypercholesterolemia | LDL-R | 7 |
| Familial defective apo B100 | Apo B | 7 |
| Gain-of-function PCSK9 mutations | PCSK9 | 7 |
| Autosomal recessive hypercholesterolemia | ARH | 7 |
| Abetalipoproteinemia | MTP | |
| Hypobetalipoproteinemia | Apo B | |
| Familial sitosterolemia | ABCG5/ABCG8 | |
| Familial Lp(a) hyperlipoproteinemia | Apo (a) | |
| Remnant Lipoproteins | ||
| Dysbetalipoproteinemia type III | ||



