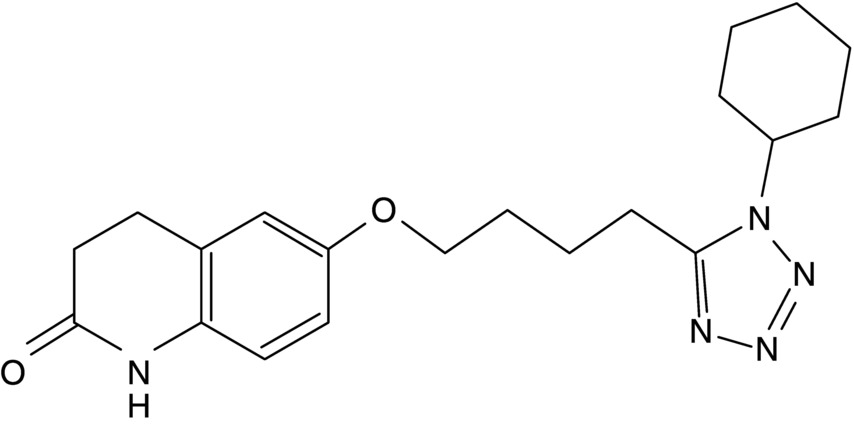
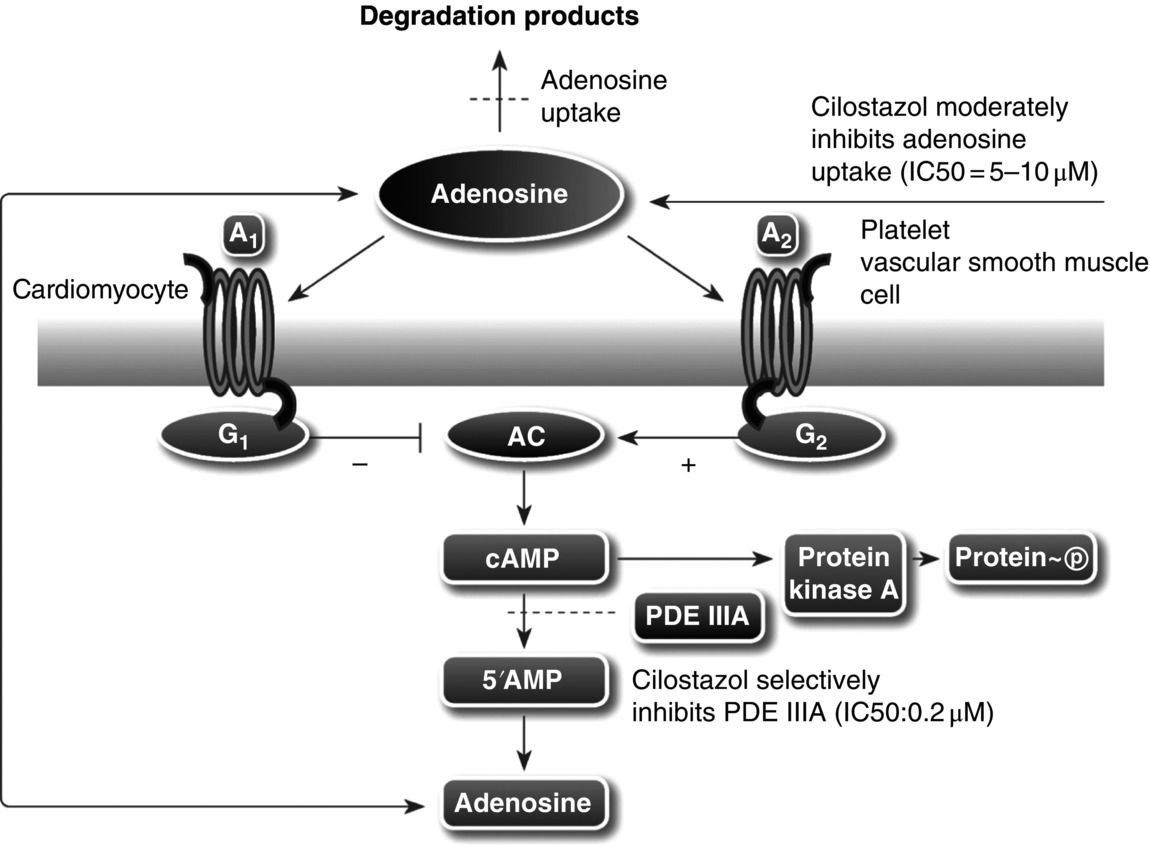
14 Seung-Whan Lee, Duk-Woo Park, and Seung-Jung Park University of Ulsan College of Medicine, Asan Medical Center, Seoul, Republic of Korea Cilostazol (6-[4-(1-cyclohexyl-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2(1H)-quinolinone; OPC-13013) (Figure 14.1) is a selective reversible inhibitor of phosphodiesterase 3 (PDE 3), resulting in increasing cyclic adenosine monophosphate (cAMP). An increased level of cAMP results in an elevation of the active form of protein kinase A (PKA), which is closely related with an inhibition in platelet aggregation. PKA also prevents the activation of an enzyme (myosin light-chain kinase) that is important in the contraction of vascular smooth muscle cells, thereby exerting its vasodilatory effect. This chapter reviews basic pharmacological actions of cilostazol that might be related to clinical efficacy. Figure 14.1 Chemical structure of cilostazol. Cilostazol is a selective, reversible phosphodiesterase 3A (PDE 3A) inhibitor with unique antithrombotic and vasodilatory properties. Cilostazol is approved for the treatment of intermittent claudication by the US Food and Drug Administration. The cilostazol does not affect other types of phosphodiesterase (PDE). However, the local tissue levels of cilostazol might be higher than the free concentration in plasma because of the lipophilicity of the drug. Meanwhile, cilostazol does not have an effect on PDE 1, 2, and 4 at comparable concentrations and only has a minor effect on PDE 5. Because PDE 3 increases the breakdown of cAMP, the inhibition of PDE 3 increases the level of cAMP. Since both platelets and vascular smooth muscle cells contain PDE 3A, pharmacological actions of cilostazol such as platelet inhibition and vasodilatory effects are associated with the inhibition of PDE 3 [1]. Recently, the inhibition of adenosine uptake has been reported to be another pharmacological action of cilostazol. This leads to enhanced adenosine actions via A1 and A2 receptors. In platelets and vascular cells, A2-mediated increases in cAMP enhance the consequences of PDE inhibition, that is, result in additional increases in cAMP. In cardiocytes carrying the A1-receptor subtype, there will be a Gi-mediated inhibition of adenylate cyclase with subsequent reduction in cAMP (Figure 14.2). Moreover, cilostazol is a unique agent that operates through its action as endothelium-targeted antithrombotic therapy, achieving its effects by improving endothelial cell function and reducing the number of platelets partially activated by interacting with activated endothelial cells. According to current knowledge, the actions of cilostazol that are most important for its clinical efficacy involve effects on platelets and vascular cells. Figure 14.2 Mechanism of intracellular cAMP control by adenosine and PDE in platelets, vascular smooth muscle cells, and cardiomyocytes. The cAMP levels are controlled by degradation via PDE and by biosynthesis via adenylate cyclase (AC). AC activity in turn is controlled by stimulatory (Gs) and inhibitory (Gi) G-proteins. Adenosine, derived from either cellular metabolism or extracellular sources, activates Gs via A2 receptors and Gi via A1 receptors. This results in either amplification or inhibition of AC. Cilostazol induces cAMP elevation in vascular smooth muscle cells via inhibition of PDE 3-dependent degradation and adenosine (A2)-induced stimulation of cAMP formation (Figure 14.2) [2]. An increased level of cAMP results in an increase in the active form of PKA, which is closely related with an inhibition in platelet aggregation. PKA also prevents the activation of an enzyme (myosin light-chain kinase) that is important in the contraction of vascular smooth muscle cells, thereby exerting its vasodilatory effect. Meanwhile, elevated cAMP inhibits calcium entry and migration, proliferation, and matrix synthesis in smooth muscle cells. Many downstream pathways have been suggested based on in vitro studies, including inhibition of [3H] thymidine incorporation into deoxyribonucleic acid (DNA), inhibition of interleukin-6 production, activation of cAMP-dependent PKA, induction of the p53 and p21 antioncogenes and apoptosis, downregulation of p27Kip1, and inhibition of expression of the E2F1, E2F2, and E2F4 proteins [3, 4]. After stent placement, cilostazol inhibits stent-induced P-selectin expression on platelets and upregulates leukocyte Mac-1, thereby inhibiting neointimal growth [5]. In the Cilostazol for REStenosis Trial (CREST), cilostazol showed reduced neointimal hyperplasia and restenosis rate after bare-metal stent implantation [6]. Cilostazol has also been shown to reduce the rate of ISR after drug-eluting stent (DES) implantation. In a group at risk of restenosis (diabetic patients, long-lesion patients), cilostazol was shown to reduce the rate of angiographic restenosis and target lesion revascularization without an increase in severe adverse effects after DES implantation [7, 8]. A recent meta-analysis of the effects of cilostazol in patients treated with both bare-metal stents and DES found that in 2809 patients pooled from 10 different randomized trials, cilostazol reduced late loss by a mean difference of 0.15 mm. Binary angiographic restenosis was also significantly lower in patients treated with cilostazol, regardless of the type of stent used [9]. Cilostazol induces elevation of endothelial cAMP via inhibition of PDE 3 degradation and A2-induced stimulation of cAMP formation in endothelial cells. Elevated cAMP stimulates endothelial cell proliferation, reduces expression of adhesion molecules (i.e., vascular cell adhesion molecule-1), inhibits cytokine release and action (monocyte chemoattractant protein-1, platelet-derived growth factor, tumor necrosis factor-α), and inhibits apoptosis; thereby, cilostazol improves endothelial cell function and reduces the number of platelets partially activated by interaction with activated endothelial cells [10, 11]. Furthermore, cilostazol induces nitric oxide (NO) production through endothelial nitric activation via a cAMP/PKA- and PI3K/Akt-dependent mechanism, and this effect is involved in capillary-like tube formation in human aortic endothelial cells [12]. Moreover, cilostazol has a potential anti-inflammatory effect on monocyte–endothelial interactions via upregulation of intracellular cAMP.
Cilostazol
Introduction
Mode of action of cilostazol
Vascular smooth muscle cells
Endothelial cells
Platelets
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


