Fig. 8.1
Baroreflex changes with sleep in one subject. Each line represents one baroreflex sensitivity estimation. A average systolic pressure awake, both before and after sleep, S average systolic pressure during slow wave sleep (Reproduced with permission from Ref. [12])
Although the neural mechanisms involved in the complex integration between sleep stages and cardiovascular reflexes are not completely known, the available evidence indicates that NREM sleep is characterized by a progressive decrease in neural sympathetic nerve activity [10] with an accompanying parasympathetic predominance [6, 11], and a downward resetting of the arterial baroreceptor reflex [12, 13]. The relative autonomic stability of NREM sleep is, however, interrupted by those burst of sympathetic nerve activity that are observed on the occurrence of arousals [10]. The resulting phasic increase in arterial pressure does not evoke the bradycardic response which would be expected due to the arterial baroreflex, but rather an increase in heart rate.
A marked sympathetic activation and instability in blood pressure and heart rate, likely of central origin, also characterize phasic REM sleep, during which sudden bursts of sympathetic nerve traffic, about twice the level seen during wakefulness, are documented [10]. As these increases in sympathetic activity are interspersed between the predominantly parasympathetic activity of tonic REM periods, this sleep stage as a whole appears to induce complex fluctuations in autonomic function.
In light of the lengthening of REM cycles as sleep progresses, it has been hypothesized that REM sleep represents a vulnerable state for the cardiovascular system that can set the stage for nocturnal arrhythmias and cardiovascular events [10, 14, 15]. The fluctuations in autonomic activity across sleep stages and their impact on arrhythmia susceptibility may become particularly dangerous in the context of underlying cardiovascular diseases that are often associated with a deranged sympatho-vagal balance [16]. It has been described that after myocardial infarction, the loss in the capability of the vagal activity to physiologically increase during sleep results in a condition of relative sympathetic dominance even during NREM sleep, normally described as a condition at low risk for lethal events [17]. However, it is the sympathetic surge of the normal morning transition from sleep to wakefulness that has been more consistently implicated in the early morning peak in the occurrence of sudden death and implanted defibrillator discharges [18, 19].
8.3 The Respiratory System During Sleep and Sleep Apnea
Falling asleep removes postural muscle tone, voluntary respiratory control, and the wakefulness stimulus for breathing, leaving metabolic demand as the primary determinant of minute ventilation [20].
The sensors for chemical stimuli include the central and peripheral chemoreceptors. With increasing hypoxemia and hypercapnia, the carotid bodies send afferent impulses to the medullary centers to increase ventilation. Similarly, with hypercapnia, linear changes in ventilation occur in response to central chemoreceptor stimulation. The control of ventilation is organized as a negative feedback system to maintain the PaCO2 at approximately 40 mmHg during wakefulness. Three main factors determine the stability of this control system [21, 22]: controller gain (the ventilatory response to changes in arterial blood gas tensions), plant gain (the responsiveness of changes in arterial blood gases to changes in ventilation), and system delays (the combined effect of circulation time from the lungs to chemoreceptors and of phase shifts resulting from mixing and diffusion processes in the lungs, heart, vasculature, and brain tissues). In general, both the hypoxic and hypercapnic ventilatory responses – that represent the important elements of the controller gain in the respiratory control loop – decline during NREM sleep and decline further with REM sleep [23]. Thus, the transition from wakefulness to NREM sleep is accompanied by a decrease in ventilation (largely through a decrease in tidal volume) associated with an increase in upper airway resistance. As a result, the sleep eupneic PaCO2 increases by 2–7 mmHg, and oxygen saturation decreases by 1–3 %. While reductions in PaCO2 during wakefulness have no or limited effects on the breathing pattern, in NREM sleep small transient reductions in PaCO2 (even only to the waking levels) result in significant apnea [24]. Breathing during REM sleep is characteristically more rapid than during NREM sleep (despite muscle atonia) as a consequence of an excitatory drive to breath. Periods of hyperventilation are interspersed with period of more regular respiration and apneas of varying length.
In most healthy subjects of any age, the loss of wakefulness influences on neurochemical control of breathing and airway patency during sleep is of minor physiological consequences. In some other healthy subjects, and even more in individuals with cardiac abnormalities, these changes predispose to respiratory abnormalities during sleep, generally referred to as sleep-disordered breathing. Commonly, sleep-disordered breathing is divided into “central,” denoting an absence or marked reduction in central motor drive to respiratory muscle and “obstructive” events which are comprised of respiratory efforts against a narrowed or closed upper airway.
8.4 Sleep Apnea in Heart Failure
Both obstructive sleep apnea (OSA) and central sleep apnea (CSA) are a common comorbidity in patients with heart failure (independently of reduced or preserved left ventricular function) (Figs. 8.2 and 8.3), occurring approximately in half of them despite optimal pharmacological therapy [25–27].
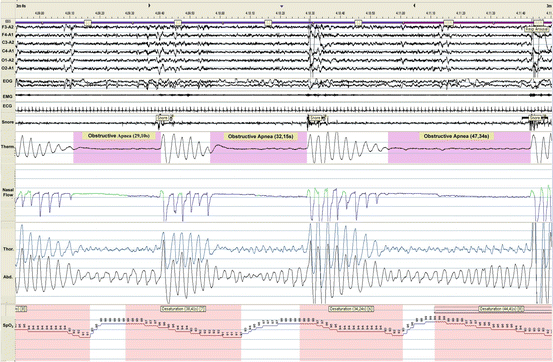
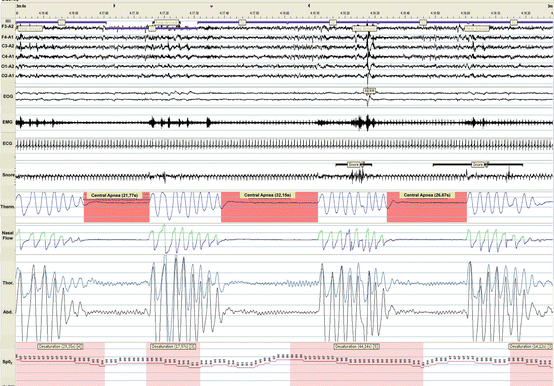
Fig. 8.2
A 3-min polysomnogram of a patient with heart failure revealing three obstructive apneas during stage 2 NREM sleep and REM sleep. Note the futile respiratory efforts associated with hypoxemia (min SpO2, 87 %) terminated by an abrupt arousal. Abbreviations: F3-A2, F4-A1, C3-A2, C4-A1, O1-A2, O2-A1, electroencephalograms, EOG electro-oculogram, EMG chin electromyogram, ECG electrocardiogram, Therm thermistor, Thor thorax, Abd abdomen
Fig. 8.3
A 3-min polysomnogram of a patient with heart failure revealing three central apneas during stage 2 NREM sleep. Abbreviations as in Fig. 8.2
Irrespective of their origin, central or obstructive, such disorders typically manifest as a cyclic waxing and waning of tidal volume [28, 29], a pattern commonly referred to as periodic breathing [28].
Using an apnea-hypopnea index (AHI, the number of apneas or hypopneas per hour of sleep) ≥15/h as a diagnostic threshold, the prevalence of OSA and CSA in patients with chronic stable heart failure and reduced ejection fraction ranges from 15 % to 26 % [25–27] and from 21 % to 38 % [25–27], respectively.
As for the relative distribution of OSA and CSA, data from previous investigations range from 28 % vs. 72 % [25, 26, 30] to 55 % vs. 45 % [27], respectively. However, it has to be underscored that many of these patients experience during the same night varying degrees of both central and obstructive sleep apneas [31]. Compared to patients without sleep apnea or OSA, patients with CSA have a worse hemodynamic profile and higher NYHA class [32].
It is recognized that heart failure contributes to the development of CSA and OSA by several mechanisms. First, in patients with heart failure, the ventilatory control system may become unstable and give rise to self-sustaining ventilatory oscillations owing to a long circulatory delay and an increased loop gain brought about, respectively, by impaired hemodynamics and augmented chemosensitivity [33, 34]. Second, the presence of a narrowed CO2 reserve enhances the susceptibility to central apneas [35]. Third, overnight rostral fluid displacement from the legs may cause fluid accumulation in the neck and lungs, thereby predisposing to the occurrence of OSA (due to the increased collapsibility of upper airways) and CSA (due to an increase in pulmonary congestion leading to worsening of the lung function and to stimulation of pulmonary vagal receptors, both of which would promote ventilatory instability) [36, 37].
The hallmark of sleep-disordered breathing – being central or obstructive in nature – is the triggering of intermittent episodes of hypoxemia and hypercapnia and of arousals which cause fragmentation of physiological sleep [38]. These events take place in repetitive cycles during sleep and induce the activation of a number of pathophysiological consequences including sympathetic activation, endothelial dysfunction, oxidative stress, metabolic dysregulation, and inflammation [39, 40], which have been shown to impact on the clinical outcome [41, 42]. Interestingly, in heart failure patients, disordered breathing also occurs during day time, possibly contributing to poor prognosis [43].
8.5 Sleep Apnea and the Autonomic Nervous System in Heart Failure
Autonomic cardiovascular control may be importantly implicated as a potential link between sleep-disordered breathing and its pathophysiological consequences. Autonomic responses to apnea are complex and mainly involve chemoreflex and baroreflex regulation.
In OSA patients, several studies have demonstrated that recurrent nocturnal apneas are followed by sympathetic activation [44] and impaired baroreflex control [45] that persist during daytime wakefulness [44, 46], thus leading to a chronic dysfunction in autonomic balance (Fig. 8.4). Interestingly, the OSA-induced baroreflex dysfunction can be partly reversed following effective continuous positive airway (CPAP) treatment [47].
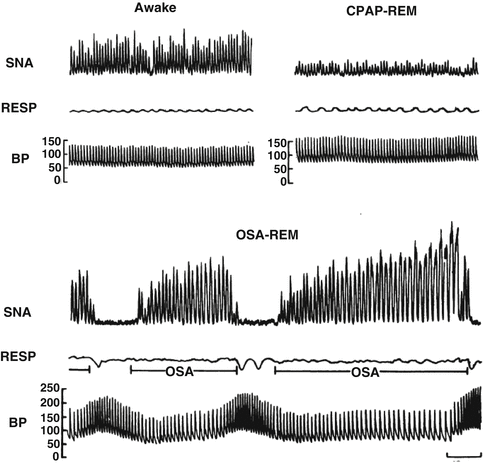
Fig. 8.4
Recording of sympathetic neural activity (SNA), respiration (RESP), and intra-arterial blood pressure (BP) in the same subject when awake, with obstructive sleep apnea during REM sleep and with elimination of obstructive apnea by CPAP therapy during REM sleep. SNA is very high during wakefulness, but increases even further secondary to obstructive sleep apnea during REM. Blood pressure increases from 130/65 when awake to 256/110 at the end of the apnea. Elimination of apneas by CPAP results in decreased nerve activity and prevents BP surges during REM sleep (Reproduced with permission from Ref. [44])
It is currently thought that in patients with heart failure, sleep-disordered breathing causes cyclical increases in sympathetic nerve activity which results from the complex interplay of several mechanisms, including: (i) the reflex cardiac response to cyclical stimulation of chemoreceptors by hypoxia and hypercapnia, (ii) central effects of the cyclical increase in inspiratory drive and of arousals from sleep on sympathetic outflow, and (iii) the elimination of reflex inhibition of sympathetic outflow by pulmonary stretch receptors during obstructive apneas [37, 48, 49].
In patients with heart failure, the sympathetic activation that accompanies sleep-disordered breathing may significantly add to the already existing autonomic dysfunction that characterizes heart failure [50], thus contributing to the progression of the disease. Not only awake sympathetic activity – as assessed by microneurography – was significantly higher in patients with heart failure and either obstructive or central sleep apnea [51], but also baroreceptor sensitivity was found to be significantly more depressed when compared to patients with similar cardiac function but without breathing disorders [52].
8.6 Sleep Apnea and Sudden Death in Heart Failure
It has been previously shown how sleep apnea can be the cause of a derangement in autonomic cardiovascular control resulting in sympathetic overactivity [44, 45]. Moreover, the autonomic effects of arousals that can occur hundreds of times per night in patients with sleep-disordered breathing, also provide the milieu for an increased arrhythmia susceptibility. Clinical data support an increased risk for nocturnal arrhythmia and sudden death in patients with sleep-disordered breathing. In the Sleep Heart Health Study, not only the prevalence of nonsustained ventricular tachycardia was significantly higher in subjects with sleep-disordered breathing as compared to those without [53], but also the relative risk of the occurrence of an arrhythmia was markedly increased within 90 s after a respiratory disturbance in subjects with sleep-disordered breathing [54]. A retrospective analysis on death certificates of patients known to have OSA demonstrated a shift to nighttime (from midnight to 6 am) in the circadian distribution of sudden death as compared to the usual early morning peak of subjects without OSA in the general population [55]. In a large study on 10,701 consecutive adults undergoing a diagnostic polysomnogram who were followed up to 15 years, OSA predicted incident sudden cardiac death, and the magnitude of risk was predicted by multiple parameters characterizing OSA severity [56].
The role of the autonomic nervous system in triggering cardiac arrhythmias might be particularly relevant with regard to arrhythmic events taking place in patients with heart failure and sleep-disordered breathing. Several studies in patients with heart failure reported that the presence of both obstructive and central sleep apnea poses an independent risk factor for the occurrence of malignant arrhythmias documented by implantable cardioverter defibrillator therapies [57, 58]. In a study analyzing the circadian pattern, a striking increase in the incidence of ventricular arrhythmias occurring during the sleep period as opposed to periods of wakefulness was observed in patients with sleep-disordered breathing [59] (Fig. 8.5). Finally, a major impairment of the autonomic control has also been documented in patients with SDB and ventricular arrhythmias suggesting that sympathetic overactivity and vagal withdrawal are important contributing mechanisms [60].
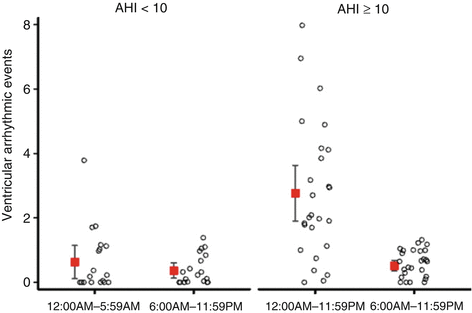
Fig. 8.5
Ventricular arrhythmias in patients with (right) and those without (left) sleep-disordered breathing according to the time of day. Error bars represent mean and 95 % confidence interval. Each circle represents the number of events in one patient. AHI apnea-hypopnea index (Reproduced with permission from Ref. [59])
8.7 Conclusions
The autonomic nervous system is deeply involved in the mechanisms mediating the cardiovascular effects of sleep within its different stages. Autonomic cardiac control during sleep is characterized by a vagal predominance for most of the time that is interrupted by bursts of sympathetic activity during arousals in NREM sleep and even more during the phasic REM sleep periods. The autonomic fluctuation during sleep associated with the lasting sympathetic activation that occurs in many cardiovascular diseases, particularly heart failure, sets the stage for an increased susceptibility to life-threatening arrhythmias. Sleep apneas, which are a common comorbidity in patients with heart failure, further complicate the picture of autonomic dysregulation and the risk of cardiac events in these subjects by eliciting a further sympathetic activation mostly due to the associated oxygen desaturation and recurrent arousals. Further understanding of the pathophysiological pathways linking sleep disorders to cardiac events is however important to improve clinical management and reduce cardiovascular risk.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


