Fig. 20.1
(a) BMS healing in animals vs. humans. Line plot showing the temporal relation of peak neointimal growth in animals and humans following the placement of bare stainless steel stent. The plots are predominantly derived from morphometric analysis of pig and human coronary stents. In animals, peak neointimal growth in stainless steel stents is observed at 28 days, compared with 6–12 months in humans (Reproduced from Virmani et al. [31], with permission of BMJ Publishing Group Ltd). (b) Arterial healing following balloon expandable BMS implantation in human coronary arteries. (a) Platelet-rich thrombus (arrows) is identified around a stent strut (*) 1 day after the placement of Driver stent. (b) A fibrin-rich thrombus (arrowhead) is focally present around a stent strut 1 day after the placement of Express stent. (c–f) Platelet- (arrow) and fibrin- (arrowheads) rich thrombus (c) with numerous inflammatory cell infiltration around stent struts (double arrows, c–e) and focal endothelialization (f). Five days after the placement of Multi-Link Vison stent. Giant cells are occasionally observed (arrows, d). The presence of inflammatory cells consisting of neutrophils and lymphocytes (boxed area in d) are highlighted in (d and g–i). Early neointima presents 23 days after Multi-Link Vision stent placement. (g) Intimal cells within extracellular matrix seen above the stent strut. (h) KP-1 immunostaining identifying macrophages (arrows) adjacent to the strut at base of neointima. (i) Actin staining identifying smooth muscle cells close to luminal surface of neointima (arrows). (j, k). Smooth muscle cell-rich neointima in stented coronary artery (duration .6 months). Actin staining shows smooth muscle cells close to luminal surface of the neointima in (i). (a–g) are stained with haematoxylin and eosin, while (j) is stained with Movat pentachrome (Reproduced from Chaabane et al. [33], with permission of Oxford University Press)
Inflammation
Acute and chronic inflammation is observed during the process of vascular healing (Fig. 20.1b). Acute inflammatory cells (neutrophils) are associated with stent struts and were observed in 79 % of sections within 3 days, 83 % of sections in 4–11 days, 72 % of sections in 12–30 days and 0 % after 30 days. Chronic inflammatory cells (lymphocytes and macrophages) around stent struts are also seen in 82 % of sections within 3 days, 67 % of sections in 4–11 days, 97 % of sections at 12–30 days and 85 % of sections after 30 days. Multinucleated giant cells were seen in only 10 % of sections at 12–30 days and 29 % of sections after 30 days. The extent of injury and the underlying plaque morphology were associated with the extent of inflammation. The cases that have medial disruption or necrotic core disruption showed a greater inflammatory response. Overall, the extent of inflammation correlated with neointimal growth [27].
SMC and Matrix
Migration and proliferation of smooth muscle cells promote organization of thrombus. From 12 to 30 days, approximately 50 % of cases showed smooth muscle cells. After 30 days, smooth muscle cells are seen in all cases. Initially, their appearance looks rhomboid in shape that gradually re-differentiate into contractile α-actin positive smooth muscle cells. There are mainly two theories about the source of smooth muscle cells. One is from medial smooth muscle cells, the other is from circulating progenitor cells [28, 29]. Up to 18 month after implantation, the neointima is rich in smooth muscle cells, type 3 collagen, proteoglycans and hyaluronan. Thereafter, smooth muscle cells tend to be more compact and align circularly towards the lumen with little matrix that is mainly type 1 collagen [30].
Endothelialization
The endothelium is denuded by ballooning and stenting. In the process of healing, neointimal coverage occurs gradually with 30 % of the stent surface area covered by endothelial cells at 1 month, and 80–100 % by 3–4 months following BMS deployment [14]. There are mainly two theories regarding the source of neointimal endothelial cells. One is from the proximal and distal non-treated segments and from branch points while the other is from circulating CD 34-positive progenitor cells. Both theories seems to contribute, however, we favor the former theory simply because re-endothelialization is greater in the proximal and distal ends than in the middle. This indicates that avoiding geographic miss (no injury to the non-stented segments) is important for re-endothelializaion to occur early [14].
Vascular Healing in Animal Models
On the other hand in animals, platelet/fibrin deposition is observed up to 3–7 days and granulation tissue is observed by 7 days consisting of smooth muscle cells, macrophages and scattered lymphocytes with luminal endothelial cells. At 14 days fibrin is still present with few chronic inflammatory cells and giant cells seen close to stent struts. At this stage smooth muscle cells within a proteoglycan matrix are observed over and between the struts, by 28-days the neointima contains a large number of smooth muscle cells, proteoglycans and collagen [31].
Mature Endothelial Cell
Mature endothelial cells have a complex set of junction proteins that help maintain the integrity of the endothelium that play a pivotal role in the regulation of its permeability and signal transduction. The endothelium provides vital anti-thrombotic and vasoprotective effects via the production of nitric oxide, prostacyclin, tissue plasminogen activator, thrombomodulin, heparin-like molecules, and tissue factor pathway inhibitor. Therefore, mature endothelial should be the ideal goal of re-endothelialization following stent placement. However, in the real world, this goal may not be achievable as the underlying plaque as well as risk factors in the individual patients may also play an important role [32].
Drug Eluting Stents – First, Second, and Third Generation
Prior to the introduction of DES, the concept of oral drug delivery to alter the neointimal response in the era of balloon angioplasty had failed, likely because our understanding of the mechanisms of neointimal growth was incomplete. We now know that smooth muscle cell proliferation and migration, along with the extent of thrombus, inflammation and injury are all important and that it is vital not to ignore any one of them. Although many cytokines and mediators are involved, the final common route is the entry of smooth muscle cells into the cell cycle pathway. Once activated the smooth muscle cells enter the cycle of cell division (G0, G1, S, G2 and M) where many factors control the rate and extent of proliferation, including cyclins that are required at different steps of the cell cycle along with cytokines and growth factors that control intracellular dependent phosphoinositide-dependent kinases and Akt. The latter is capable of phosphorylating the mammalian target of rapamycin [33] (Fig. 20.2).
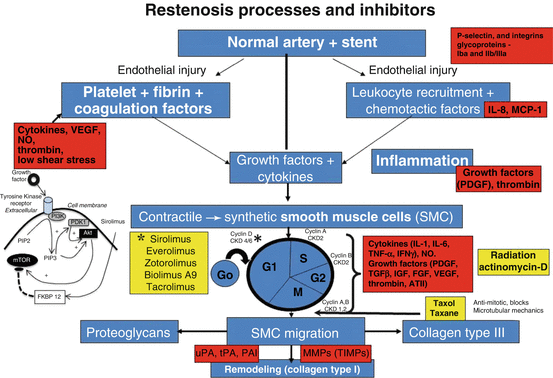
Fig. 20.2
Overview of the molecular mechanisms of restenosis and their inhibitors. The normal coronary artery is injured secondarily from ballooning and stenting resulting in endothelial cells (EC) loss followed by mural thrombus consisting of platelet aggregation with fibrin deposition. The platelets within the thrombus release a number of chemokines essential for the initiation of inflammation. This includes activation of P-selectin and integrins such as b2-integrin Mac-1 and thrombin. Local production of chemokines like interleukin (IL)-1, IL-6, interferon (IFNγ), and tumor necrosis factor-alpha (TNFα) by macrophages result in the induction of inflammation at the injury site. The injury is not only to the endothelium but also to the underlying medial wall. There is an imbalance between stimulatory growth factors (PDGF, FGF, TGF-β, and IGF-1) and chemokines and the inhibitory factors like endothelial-derived nitric oxide and heparin sulfate proteoglycans that result in the activation of smooth muscle cells (SMCs). The SMCs within the media transform from a quiescent contractile to a synthetic cell which not only proliferate but also migrate into the intima. Within the intima they further proliferate but also secrete extracellular matrix. SMC proliferation involves cell division with different cyclins that are required at different steps of the cell cycle. CDK (cyclin-dependent kinase) activity is also regulated by cell-cycle inhibitory proteins called cyclin-dependent kinase inhibitors (CKIs), which counteract CDK activity. SMC membranes are also stimulated by several classes of tyrosine kinase receptors (TKR). One of the intracellular signalling pathways involves the phosphatidylinositol (PI)3-kinase (PI3K) pathway. PI3K is a lipid kinase that phosphorylates PI. The dominant lipid product generated by PI3K—PI 3,4-biphosphate (PIP2) and PI 3,4,5-triphosphate (PIP3), the latter recruits phosphoinositide-dependent kinase-1 and Akt. Akt is then capable of phosphorylating the mammalian target of rapamycin (mTOR). The yellow squares show the various inhibitors and at which site of the cell cycle they are involved, especially sirolimus and its analogues which inhibit mTOR, whereas paclitaxel blocks microtubular mechanisms (Chaabane et al. [33], with permission of Oxford University Press, and Charron et al. [49])
The better understanding of the importance of SMC proliferation and migration resulted in the use of the immunosuppressant drug, like sirolimus to not only affect SMC and endothelial cell proliferation but also to decrease inflammation [33]. However, the cytotoxic drug, paclitaxel was also used in the first generation of drug eluting stents. In order to deliver drug, anywhere from 1 to 3 months and longer, it required the use of a polymer coating, which in the first and second generation have been permanent, whereas in the third generation, they are biodegradable polymers as the polymer disappears once the drug is delivered (Fig. 20.2). Clinical studies as well as autopsy studies have shown a marked reduction in neointimal formation with DES, irrespective of strut size, using permanent or bioabsorbable polymers. Most have used sirolimus or its analogs as drugs of choice and the results in both early and long-term clinical trials have been better with sirolimus analogs than with cytotoxic drugs, like paclitaxel [34]. All have shown delayed vascular healing as the mechanism of reduced neointima growth [14].
Delayed Healing in DES
We have shown in both the first and second generation DES that there is delay in vessel healing as compared to BMS (Fig. 20.3). We defined delayed healing as the persistence of fibrin in animal models at 28-days along with poor smooth muscle cell presence and incomplete endothelialization. However, animal models have variability with complete endothelialization observed in the porcine coronary model at 28 days although fibrin deposition persists, whereas there is greater delay in endothelialization in the rabbit iliac artery model with incomplete endothelialization observed even at 28-days. In man, complete endothelialization is observed at 3–4 months in BMS but in DES there is a delay which persists for a long time [14]. In a human autopsy study [14], when comparing first generation DES to BMS stents that had been implanted >30 days (implant duration 223 ± 253 and 229 ± 360 days, respectively) DES showed greater suppression of neointimal growth than BMS (neointimal area 2.8 ± 1.1 vs. 4.9 ± 3.0 mm2, p < 0.0003). On the other hand, covered struts (% strut endothelialized) were significantly delayed in DES than BMS (55.8 ± 26.5 vs. 89.8 ± 20.9 %, p = 0.0001, respectively) and fibrin deposition was greater in DES than BMS (fibrin score 2.3 ± 1.1 vs. 0.9 ± 0.8, p < 0.0001, respectively). Therefore, it is not surprising in our study that late stent thrombosis in the first generation DES was significantly higher as compared to BMS (61 % vs. 8 %, respectively, p = 0.0001) [14]. In a detailed study of examining only DES, 28 with thrombosis and 34 without thrombosis, those with thrombosis had a lower percentage of endothelialization coverage as compared to those without thrombosis (40.5 ± 29.8 vs. 80.0 ± 25.2 %, P < 0.0001) [35]. The ratio of uncovered to total stent struts (RUTSS) per section was also significantly greater in thrombosed DES lesion than non-thrombosed DES lesion (0.50 ± 0.23 vs. 0.19 ± 0.25, P < 0.0001). What is interesting is that the distance between individual stent struts (inter-strut distance) was significantly less in thrombosed DES lesions than non-thrombosed DES lesions (0.52 ± 0.24 vs. 0.70 ± 0.25 mm) suggesting that regional drug concentrations determine strut coverage and vessel healing rates. Endothelialization was the best predictor of late stent thrombosis by multivariate logistic analysis. The odds ratio for thrombosis was nine times greater when >30 % uncoverd struts were present as compared to those with <30 % [35]. Recently introduced intravascular imaging technologies such as optical coherence tomography (OCT) and optical frequency domain imaging (OFDI) can recognize uncovered struts and have been shown to correlate with stent thrombosis [36].
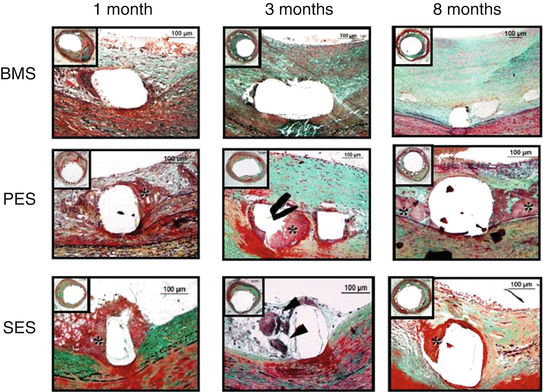
Fig. 20.3
Delayed arterial healing following drug-eluting stent (DES) versus bare metal stent (BMS) implantation. Time course of arterial healing in BMS, paclitaxel-eluting stents (PES: Taxus DES), and sirolimus-eluting stents (SES: Cypher DES) from 1 to 8 months after stent implantation. Although some peristrut inflammation is observed in BMS at 1 month, complete arterial healing, including a well-established neointimal layer, is seen at 3 and 8 months’ duration. PES shows early fibrin deposition surrounding stent struts (*), which persists up to 8 months, as a sign of delayed healing. In contrast, SES shows predominance of inflammatory cells, including giant cell formation (black arrowheads), at early time points (1 and 3 months), whereas fibrin deposition is stronger at 8 months (Reproduced from Lüscher et al. [50], with permission of Wolters-Kluwer Health)
Acute Myocardial Infarction
Underlying plaque morphology also plays an important role in the process of vascular healing after DES implantation. Ruptured plaque and strut penetration into a necrotic core have been reported to trigger restenosis in BMS and delayed vascular healing in DES (Fig. 20.4). The culprit site of acute myocardial infarction (AMI) showed delayed healing compared to the non-culprit site within the same lesion [37]. The culprit site showed greater RUTSS (49 % vs. 9 %, P = 0.01), greater fibrin deposition (63 ± 28 % vs. 36 ± 27 %, P = 0.008) and a greater inflammation score (35 % vs. 17 %, P = 0.003) than the non-culprit site. What is interesting is that neointimal thickness at the culprit site was significantly less than at the non-culprit site (0.04 vs. 0.07, P = 0.008). Current DESs (-limus and paclitaxel) are highly lipophilic drugs and therefore it is possible that these drugs have a high affinity for lipidic necrotic core and dwell within these areas for a longer period. It is likely that culprit sites with necrotic core and persistent drug show greater delayed healing than non-culprit sites with less drug persistence because of underlying fibrous or calcific plaques.
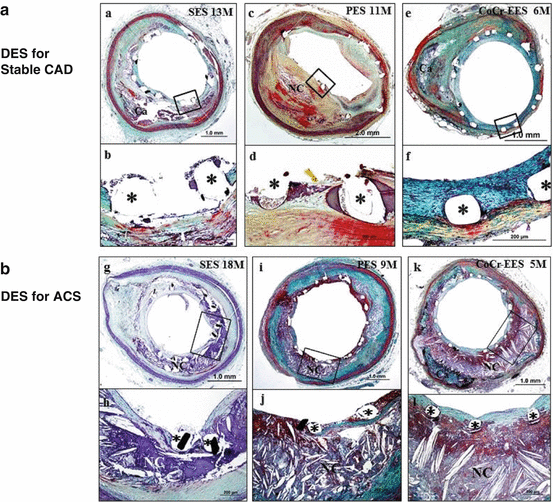




Fig. 20.4
DES for stable ACS and DES for ACS. Representative images of sirolimus-eluting stent (SES), paclitaxel-eluting stent (PES), and cobalt-chromium everolimus-eluting stent (CoCr-EES) implanted for stable coronary artery disease (CAD; a: a–f) and for acute coronary syndromes (ACS; b: g–l). a and b, Histological sections from a 53-year-old-man with SES implanted in the proximal left anterior descending coronary artery for 13 months. A low-power image (a) shows mild neointimal growth and underlying fibrocalcific plaque. Focal uncovered struts are highlighted in a high-power image in b.*Stent strut. c, d histological sections from a 71-year-old man with PES implanted in the right coronary artery 11 months antemortem. A low-power image (c) shows mild to moderate neointimal proliferation and underlying fibroatheroma. Note uncovered struts with persistent peri-strut fibrin deposition shown at high-power image in d. e, f histological sections from a 60-year-old man who received CoCr-EES in the mid left circumflex artery 6 months antemortem. A low-power image (e) shows mild neointimal proliferation and underlying fibrocalcific plaque. All struts are covered with proteoglycan-rich neointima with absence of fibrin, which is highlighted in a high-power image in f. g, h histological sections from a 74-year-old woman who received SES in the proximal left anterior descending coronary artery for acute myocardial infarction 18 months antemortem, who died of diffuse severe coronary artery disease. A low-power image (g) shows mild neointimal proliferation. Note focal uncovered struts and strut penetration into the necrotic core (NC; h). i and j, Histological sections from a 64-year-old woman with PES implanted in the right coronary artery for acute myocardial infarction 9 months antemortem, who died of congestive heart failure. A low-power image (i) shows patent lumen with stent struts surrounded by fibrin and an underlying NC. Note uncovered struts with fibrin deposition that overlie the NC (j). k and l, Histological sections from a 67-year-old man who received CoCr-EES in the proximal left anterior descending coronary artery for non–ST segment elevation acute myocardial infarction 5 months antemortem, who died of noncardiac causes. A low-power image (k) shows mild neointimal proliferation and an underlying large NC. All struts are covered with a thin neointima overlying the NC, which is highlighted in the high-power image in l. All histological sections are stained with Movat pentachrome. DES indicates drug-eluting stents (Reproduced from Otsuka et al. [16], with permission of Wolters-Kluwer Health)
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
