Figure 23–1.
Differences between decompensated and compensated hypertrophy. (a) Evolvement of cardiac output over time in compensated (grey line) and decompensated (yellow and red lines) hypertrophy after an initiating event. (b) In compensated hypertrophy wall thickness and luminal volume increase (grey circle), while in decompensated hypertrophy dilatation occurs (red circle). (c) Lower part: the differences in intracellular calcium levels at baseline (black) and in compensated (grey) and decompensated (red) hearts. In the upper part the accompanying cardiac action potentials are shown for reference
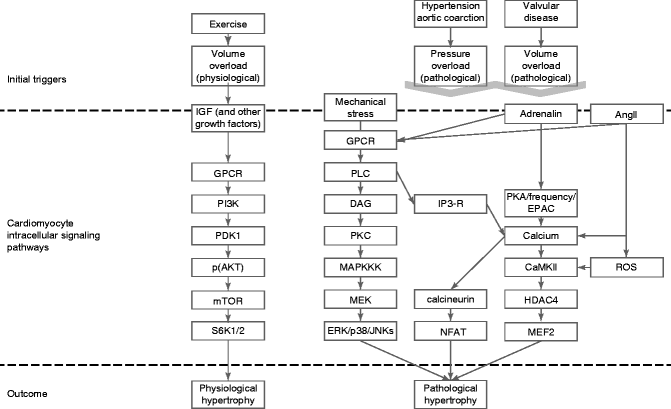
Figure 23–2.
The intracellular signal transduction pathways involved in cardiac hypertrophy leading to either a more physiological or pathological phenotype
Ventricular remodeling is known to increase the propensity for ventricular arrhythmias and may lead to sudden cardiac death. Despite a clear association between left ventricular mechanical dysfunction, hypertrophy and ventricular arrhythmias [16], the majority of sudden cardiac death occurs at earlier stages of the disease process, even in circumstances in which mechanical dysfunction or ventricular hypertrophy, are absent [16– 18]. The mechanisms by which contractile and electrical remodeling predispose to arrhythmias remain unclear, but the changes in [Ca2+]i handling and repolarization as seen in the compensated form (Fig. 23.1c) may contribute. Therefore, in this chapter, we will concentrate on a canine model of complete atrio-ventricular block (CAVB) in which the beneficial adaptations leading to compensated biventricular hypertrophy are accompanied by a detrimental one, being an increased susceptibility for repolarization related ventricular arrhythmias. We will discuss the specific remodeling changes that are related to this phenotype.
Ventricular Remodeling in the CAVB Dog
Creation of chronic CAVB by radiofrequency ablation results in numerous adaptations initiated to overcome, acutely and in the long run (weeks), the abrupt decrease in cardiac output (Fig. 23.3a, black line) due to the bradycardia (Fig. 23.3a, red line). The CAVB dog [19] is a model of compensated biventricular hypertrophy with a long QT phenotype [3, 6, 19, 20]. The primary compensatory parameter is illustrated in the recovery of cardiac output after the initial decline. Initially, this is reached by increasing cardiac contractility as can be seen in left ventricular (LV) pressure development over time (LV dP/dt, Fig. 23.3a, green line). However, as time progresses, a decline is seen, until it reaches a stable state at >10 weeks that accounts for 120 % of baseline values. Nonetheless, also on this longer time scale cardiac output is maintained, because biventricular hypertrophy develops (increased heart weight to body weight, Fig. 23.3a, yellow line) creating a new equilibrium between wall thickness and LV volume, which prevents deterioration into dilated cardiomyopathy (for reference: see Fig. 23.1) [3, 21].
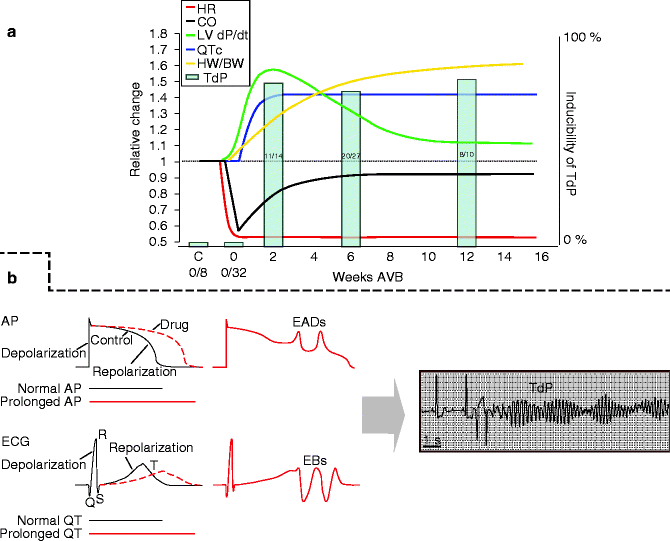
Figure 23–3.
Progress of remodeling in the CAVB dog. (a) Schematic figure summarizing the development of cardiac remodeling in the CAVB dog over time. Scale is relative. The red line shows development of heart rate. The black line is an approximation of cardiac output. The green line shows contractile remodeling as left ventricular pressure changes. The yellow line represents structural remodeling, via cardiac hypertrophy as heart weight to body weight. The blue line depicts QT interval lengthening on the ECG, as a parameter for electrical remodeling. Finally, the bars represent fraction of dogs sensitive to drug-induced TdPs (see right Y-axis) at 2, 6, or 12 weeks of chronic AV-block. (b) Summary of the mechanism of drug-induced repolarization-dependent arrhythmias. From lengthening of the QT interval/APD, to early afterdepolarizations (EADs) and extra beats (EBs), to Torsade de Pointes arrhythmias (TdP)
Aside from the structural and mechanical changes, electrical remodeling is also present. Grossly, this is identified in a lengthening of the cellular action potential duration, that in vivo is reflected in prolongation of the QT-time (Fig. 23.1, blue line) [3, 7, 17, 19–21]. This is pro-arrhythmogenic, as further drug-induced lengthening leads to early afterdepolarizations (EADs), extra beats, and ultimately life threatening Torsade de Pointes arrhythmias (TdP) (Fig. 23.3b) [17, 19, 22–24].
In the following paragraphs, we will describe in depth the electrical, contractile, and structural remodeling. Moreover, the possible mechanisms responsible for the enhanced arrhythmogeneity (arrhythmogenic outcome) and possible responsible intracellular signaling pathways will be discussed. Special focus will be on the existing intricate connections between ventricular remodeling and their effect on arrhythmogeneity.
Electrical Remodeling
As mentioned above, the most striking electrical remodeling is seen in a lengthening of the action potential duration, which can be observed via lengthening of the QT interval on the ECG or, more regionally, via an increase in the duration of the catheter-based monophasic action potentials (MAPD, Table 23.1) [17, 19, 22, 24] or the activation recovery interval (ARI) of a needle measurement [25]. The prolongation of the MAPD is larger in the LV versus the right ventricle (RV), indicating that interventricular dispersion assessed as LV MAPD – RV MAPD is increasing too (Table 23.1) [17, 19, 24, 25]. More recently, spatial dispersion was further assessed using 66 needles with 4 electrodes, demonstrating increases in transseptal, interventricular, LV transmural as LV apex to base dispersion [25]. Also temporal dispersion, quantified as short term variability of beat-to-beat repolarization was increased after CAVB [24]. So not only is the repolarization prolonged, but it is also non-uniform in space and time.
Table 23–1.
Characterization of electrical remodeling in the CAVB dog: summary of studies which report APD lengthening and variability, via monophasic action potential duration (MAPD) or QT interval lengthening on the ECG in AV-block dogs
Reference | QT | MAPD | Spatial dispersion | Temporal dispersion |
|---|---|---|---|---|
Vos et al. [19] | ↑ | ↑ | ↑ | |
Schoenmakers et al. [17] | ↑ | ↑ | ↑ | |
Boulaksil et al. [25] | ↑ | ↑(ARI) | ↑(ARI) | |
Thomsen et al. [24] | ↑ | ↑ | ↑ | ↑ |
Dunnink et al. [22] | ↑ | ↑ | ↑ | ↑ |
Lengthening of the QT-duration develops in the first 2 weeks after AV-block and then stabilizes (Fig. 23.3a, blue line) [23]. Cellular experiments performed at 3–9 weeks of CAVB in isolated cardiomyocytes confirm this increase in repolarization times [26, 27]. Since potassium currents are largely responsible for proper repolarization [28], the most logical assumption would be to expect a decrease in these currents in the CAVB dog. Indeed, both the slow component of the delayed rectifier current Iks, as to a lesser extend the rapid component (Ikr), are downregulated in the LV and in the RV (Table 23.2) [29]. Other repolarizing currents as the inward rectifying potassium current (Ik1) and the transient outward current (Ito) are not changed functionally in either of the two ventricles [29].
Table 23–2.
Characterization of electrical remodeling in the CAVB dog: summary of the ion current changes observed in the CAVB dog
Reference | Etiology | INa | ICaL | Ito | Ikr | Iks | Ik1 | NCX | Na/K pump | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Sipido et al. [6] | CAVB | = | = | = | ↑ | ↑ | |||||||||||
Volders et al. [29] | CAVB | =. | = | = | ↓ | ↓ | ↓ | =. | = | ||||||||
Boulaksil et al. [25] | CAVB | ↓ (peak) | = | ||||||||||||||
Antoons et al. [30] | CAVB | ↓ Late | |||||||||||||||
Antoons et al. [31] | CAVB | ↑Window current | |||||||||||||||
Stengl et al. [26] | CAVB | ↓ | |||||||||||||||
Verdonck et al. [32] | = | ||||||||||||||||
Also the inward currents are attenuated when compared to their functioning before AV-block. The peak sodium current (INa) is down in the LV (Table 23.2) [25], whereas INa late, which occurs in the plateau of the action potential, is also reduced [30]. The current through the L-type calcium current (ICa-L) does not change after AV-block [6], but a more frequent occurrence of the window current is observed [31]. Sipido et al. [6] observed an increased current via the sodium calcium exchanger (NCX), in both modes, responsible for a more active sarcolemmal exchange of calcium and natrium ions through this channel. Also this increase may be pro-arrhythmic (see further). In line with this observation, [Na+]i seems to be increased, while activity of the Na+/K+ exchanger remained similar [32]. The reduction in both peak and late sodium current are not in line with an increase in [Na+]i or [Ca2+]i nor with an increase in cellular APD. Recently we studied the relevance of the sodium-proton exchanger (Na+/H+) (van Borren et al., 2009, unpublished data). The activity of this pump seems elevated thereby possibly explaining the increased [Na+]i. Taken together, repolarization reserve in the CAVB model has been diminished.
For a complete summary of the observed changes in ion currents in the CAVB dog, see Tables 23.1 and 23.2. On mRNA and protein level, appropriate changes of the (alpha)α-subunits of ion channel proteins have been observed [26, 33]. A reference for the role of ion currents in a normal action potential can be found in Fig. 23.4a.
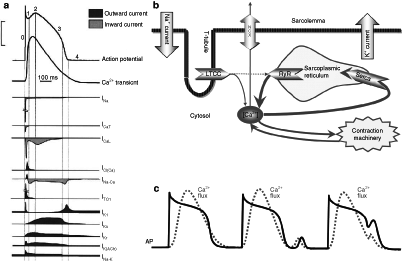
Figure 23–4.
Ion currents and calcium handling in the cardiomyocyte. (a) Schematic illustration of the depolarizing and repolarizing currents that shape the action potential in the normal mammalian ventricle. Time course of each of the currents is shown together with the course of the Ca2+ transient. (b) Depiction of intracellular calcium movement during an action potential. Upon depolarization the L-type calcium channel opens (LTCC) and a relatively small amount of calcium enters the cardiomyocyte. The ryanodine receptor (RyR) reacts to this calcium and opens as well, leading to release of calcium out of the sarcoplasmatic reticulum. The myofilaments start contracting in response to the increased calcium concentration. At the end of the action potential the intracellular calcium concentration is reduced to baseline by the sarcoplasmic reticulum calcium-ATPase (SERCA) pump, which pumps calcium back in to the sarcoplasmic reticulum, and via the sodium/calcium exchanger (NCX), which removes a smaller amount of calcium in exchange for sodium ions (three sodium ions for one calcium ion). (c) Behaviour of the intracellular calcium flux during a normal action potential, and a delayed and early afterdepolarization. The dotted line depicts the intracellular calcium concentration. Note that a delayed afterdepolarization occurs after the end of the action potential, while a early afterdepolarization occurs during the repolarization phase of an action potential
Contractile Remodeling
The ion current responsible for coupling the electrical impulse to contraction is ICaL via the L-type calcium channel (LTCC) (Fig. 23.4b) [34], which is voltage-sensitive and activates upon depolarization. The ryanodine receptor (RyR), located at the sarcoplasmic reticulum (SR), opens in accordance with the influx of calcium through LTCC, and even more calcium will be released in the cytoplasm; a process called ‘calcium-induced calcium release’ [34]. The SR functions as an intracellular calcium store. Cytosolic calcium binds to the myofilaments and myocyte shortening/contraction occurs. Thereafter [Ca2+]i returns to baseline values with the assistance of the NCX, transporting calcium out of the cell, while the (larger) remainder of cytoplasmic Ca2+ is pumped back into the SR via the sarcoplasmic reticulum calcium ATPase (SERCA). This is summarized in Fig. 23.4b and in the left part of Fig. 23.4c. Also, for a more detailed review of excitation-contraction coupling, see Bers [34].
Contractile strength can be varied under regulation of adrenergic stimuli, like epinephrine. The ß(beta)-adrenergic pathway modifies contraction by phosphorylation of a number of key proteins: (1) LTCC, (2) RyR release, and (3) SR reuptake of calcium by phosphorylation of phospholamban, the ancillary inhibitor protein of SERCA [34]. Also, CamKII has a positive, facilitating function in increasing contraction by phosphorylation of the same set of proteins, although at a different amino-acid residue [35].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


