(1)
Project-team INRIA-UPMC-CNRS REO Laboratoire Jacques-Louis Lions, CNRS UMR 7598, Université Pierre et Marie Curie, Place Jussieu 4, 75252 Paris Cedex 05, France
Abstract
Vasculogenesis, an embryological process, defines the formation of capillary plexi from endothelial precursor cells. A primitive vascular network is built via the assembly of angioblasts. Initial patterning of embryonic vascular network is independent of hemodynamic forces. The onset of blood circulation contributes to vasculature remodeling.
Vasculogenesis, an embryological process, defines the formation of capillary plexi from endothelial precursor cells. A primitive vascular network is built via the assembly of angioblasts. Initial patterning of embryonic vascular network is independent of hemodynamic forces. The onset of blood circulation contributes to vasculature remodeling.
Angiogenesis corresponds to the maturation of the primary vascular network during embryo- and fetogenesis as well as the expansion of existing vasculature, i.e., generation of new branches from this network in utero as well as vasculature extension after birth during tissue development and repair. Angiogenesis enables delivery of oxygen and nutrients toischemic regions. An intermittent, low-magnitude ischemia during exercise in normal conditions as well as during ischemic preconditioning activates angiogenesis for muscle adaptation.
Angiogenesis relies on the integration of hemodynamic and chemical signals. The vascular growth requires the coordinated proliferation and migration of endothelial cells. It involves vessel dilation or contraction, sprouting and branching, as well as intussusceptive involution and pruning (non-functional vessels). Intussusception, another type of remodeling of pre-existing vessels, i.e., insertion of tissular pillars into blood vessel lumens, enables vessel splitting.
Transcriptional, post-transcriptional, and post-translational mechanisms participate in the control of endothelial cell behavior during sprouting, branching with leading endothelial tip cells and trailing stalk cells, and tubulogenesis [886]. Specialized endothelial tip cells at the leading edge of vascular sprouts form filopodia in response to guidance signals.
Lymphangiogenesis is the development of new lymph vessels.Arteriogenesis deals with formation of mature arterioles and arteries withsmooth myocytes, particularly for collateral development to bypass an obstructed artery.
Neovascularization involves 3 major processes stimulated by several chemical, physical, and mechanical (wall shear stress and intramural [circumferential and longitudinal] tension) factors: (1) postnatal vasculogenesis, i.e., formation of new blood vessels from bone marrow-derived endothelial progenitor cells; (2) angiogenesis; and (3) arteriogenesis, i.e., growth and remodeling of arterioles into arteries (collateralization).
10.1 Vasculogenesis
Mesodermal cells in the early embryo differentiate into endothelial progenitor cells —angioblasts — and aggregate. A functional vasculature develops from angioblasts.
Duringvasculogenesis, angioblasts determine “blood pockets” that lengthen to form irregular capillaries. Vascular plexi formed by aggregating angioblasts remodel and mature into organized vascular networks of large and small, ramified or merged, vessels. Parts of vascular plexi contain blood islands. Outer cells of the blood islands give rise to endothelia, inner cells to hematopoietic progenitors [1167]. Fusion of angioblast pockets forms primary capillary plexi. Ducts connect each other in a non-hierarchical, inhomogeneous network of primitive vessels.
Immediately after vasculogenesis, angioblasts and endothelial cells undergo specification to either arterial or venous fate. Angioblasts that aggregate to form blood islands, which fuse and remodel in response to hemodynamic stresses and genetic factors,1 create a primitive interlaced network of arterial and venous plexi.
Angioblasts also acquire arterial or venous fates and coalesce to generate the first embryonic blood vessels: the dorsal aorta and cardinal vein. The dorsal aorta and cardinal vein are indeed directly formed by the coordinated sorting and segregation of arterial and venous angioblasts and their respective assembly.
After vasculogenesis and once associated with the heart pump, the primitive network that conveys blood remodels with branching. Angiogenesis from the dorsal aorta, cardinal vein, and vascular plexi builds a hierarchical network of arteries, arterioles, capillaries, venules, and veins. Subsequent recruitment pericytes and vascular smooth myocytes stabilizes nascent vessels and promotes vessel maturation. In addition, sprouting of lymphatic endothelial cells from venous endothelia gives rise to the closed-ended lymphatic network. Furthermore, a subset of hemogenic arterial endothelial cells generates hematopoietic stem cells.
10.2 Angiogenesis
Angiogenesis is the development of new branching vessels from existing vasculature. Angiogenesis thus involves migration and proliferation of endothelial cells from pre-existing vessels. Angiogenesis includes sequential events: (1) increased capillary permeability and endothelial cell and pericyte activation and hypertrophy; (2) destabilization with degradation of the vascular basement membrane and remodeling of the extracellular matrix; (3) endothelial cell proliferation and migration in the target extracellular matrix; (4) capillary lumenogenesis (Sect. 10.2.5); and (5) maturation with recruitment of pericytes, subsequent inhibition of endothelial proliferation, basement membrane reconstitution, and junctional complex formation, that stabilizes new vessels. Bone marrow-derived pericytes are also recruited for angiogenesis, particularly after ischemia [1168]. Angioblasts are able to differentiate into both blood and endothelial cells.
Both chemotaxis and haptotaxis 2 contribute to tissue development, defense, and repair. Various fibronectin-bindingintegrin types collaborate to yield cell adhesion and migration onfibronectin [1164]. In addition, disruption of epithelial layers instantaneously generates lateral electric fields [1165], directed toward the wound center, with sustained outward electric currents. The lesion shunts the transepithelial potential difference. These electric fields trigger cell migration during wound healing. Electrotaxis is controlled by PI3Kc1γ andPTen phosphatase [1166].
Vascular sprouts develop and build a structure. Angiogenesis is tightly coupled to tissue development to supply the growing tissue with oxygen and nutrients and remove its metabolic waste. The three-dimensional, fractal-like network of branched vessels results from local growth gradients over small distances. Localized production of growth factors promotes tissue expansion and determination of the position of branching nodes. The process is adaptive, because vessel development, maturation, and regression coexist. Local control of cell responses to stimuli, such as the regulation of the extracellular matrix, avoids disorganization.
Angiogenic vessels differ from mature vessels. The wall structure is not well organized. The interactions between endothelial cells and pericytes are impaired. The wall is leaky. Angiogenic endothelial cells have altered surface markers and adhesion molecules.
Vessel maturation leads to a fully formed and functional network. After suppression of endothelial proliferation and sprouting, mural cells are incorporated into vessel walls and structural elements are constructed (valves, fenestrations or, conversely, tight junctions, etc.).
10.2.1 Stem, Progenitor, and Precursor Cells in Angiogenesis and Neovascularization
Fetal cells (trophoblasts, erythroblasts, leukocytes, hematopoietic progenitors, and mesenchymal stem cells) migrate in the maternal circulation during pregnancy. Fetal endothelial progenitors can then participate in maternal angiogenesis during pregnancy [1169]. After birth, angiogenesis participates in organ growth. During adulthood, angiogenesis in a healthy subject mainly occurs in the cycling ovary and the placenta during pregnancy. Otherwise, angiogenesis appears in trauma sites, woundhealing, and developingtumors.3
Among subsets of mesenchymal stem cells,circulating endothelial progenitors contribute to angiogenesis, even during steady-state conditions in adult humans.Circulating angiogenic cells participate in tissue repair.
The sequential recruitment of mesenchymal stem cells begins by their mobilization from the bone marrow through rupture of the CXCL12–CXCR4 complex [1170]. Their migration to injury sites results from chemotaxis, not only via the CXCL12–CXCR4 complex, but also via CCL7 chemokine that targets CCR1, CCR2, and CCR3 receptors [1170].4 Chemotaxis of hematopoietic progenitor cells and circulating angiogenic cells is also triggered by the CCL19–CCR7 complex, but the cell migration is weaker than that stimulated by CCL7 chemokine. In addition, activated macrophages produce interleukin-6 that regulates the migration of endothelial progenitor cells via interleukin-6 receptor-α chain. Chemokine CX3CL1 can also be involved, as its CX3CR1 receptor resides on endothelial progenitor cells.
Bone marrow-derived endothelial progenitors are recruited totumoral growing vessels using transcriptional Class-B basic helix–loop–helix protein bHLHb24 factor. Attracted endothelial progenitors help in the progression of dormant micrometastases to lethal metastases [1171].
Recruitment of myelomonocytic cells from the bone marrow to tissues can serve as a source of pro-angiogenic cytokines afterischemia. The coexistence of myeloid lineage progenitors capable of endothelial differentiation and pro-angiogenic myeloid accessory cells then leads to 2 complementary mechanisms of angiogenesis. Vascular endothelial cells can differentiate from common myeloid progenitors and more mature granulocyte–macrophage progenitors [1172]. Bone marrow-derived progenitors of endothelial cells express PECAM1, von Willebrand factor, and TIE2, but not PTPRc, and pericyte marker desmin and smooth muscle actin.
10.2.2 Involved Factors in Angiogenesis
Numerous mechanisms are involved at various length scales: chemical signaling and genetic response, cell interactions, and environmental stresses.5 Computational simulations have been proposed to provide insights into structure–function relationships at all involved scales [1173]. Vasculogenesis is modeled as traction-driven remodeling of an initially uniform tissue in the absence of blood flow, and angiogenesis as a flow-driven remodeling of a porous structure.
Development of vascular trees includes adaptation to mural stress field. Angiogenic molecules are generated in response tohypoxia and other stimuli.Angiopoietins, VEGF, andintegrins regulate the vessel caliber. The expression of angiopoietin TIE2 receptor characterizes 3 cell types that have angiogenic activity: (1) endothelial cells; (2) TIE2 + monocytes and their hematopoietic progenitors, and (3) pericyte precursors of mesenchymal origin [1174].
10.2.3 Endothelial Sprouting: Tip and Stalk Cells
Certain endothelial cells that will form the distal end of the sprout — tip cells— strongly express vascular endothelial growth factorVEGFR2 receptor. Once endothelial cells are selected for sprouting, sprouting is controlled by the balance between angiogenic signals (e.g., VEGF and ephrin-B2; Tables 10.1 and 10.2) and antagonists (tight contacts with pericytes recruited by platelet-derived growth factor-B, certain extracellular matrix components, and VEGF inhibitors).
Table 10.1
Endothelial sprouting (Part 1); Source: [886]; DLL: Delta-like ligand; EC: endothelial cell; ECM: extracellular matrix; MMP: matrix metallopeptidase; Nrp: neuropilin; Robo: Roundabout homolog; S1P: sphingosine 1-phosphate; Sema: semaphorin (Sema, Ig, transmembrane, and short cytoplasmic domain); TIE: Tyr kinase with Ig and EGF homology domains (angiopoietin receptor); Unc5b: Uncoordinated-5B homolog; VEGF: vascular endothelial growth factor; VEGFR: VEGF receptor). Endothelial cells produce platelet-derived growth factor PDGFb, transforming growth factor TGFβ1, angiopoietin-2 (Ang2), S1P and S1P receptor S1P1, TIE2 receptor, and VEGFR1 to VEGFR3 receptors. Mural cells synthesize angiopoietin-1 (Ang1), S1P, activin receptor-like kinase ALK5, which heteromerizes with Tβ R2 receptor, and PDGFb. Signaling via VEGFR2, VEGFR3, or VEGFR2–VEGFR3 heterodimers is pro-angiogenic. Cleaved VEGFc and VEGFd interact with VEGFR2; VEGFa connects to VEGFR1 and soluble VEGFR1S (secreted extracellular domain) that then limit VEGFa availability. Vascular endothelial growth factor VEGFa and Ang2 support mural cell detachment and vessel destabilization. Receptor TIE2 tethers to matrix-associated Ang1 at adhesion sites between endothelial cells and matrix, thereby assisting migration. On the other hand, at EC–EC adhesions, the PTPRb–TIE2–Ang1–TIE2–PTPRb complex between apposed cells. In addition, Ang2 antagonizes Ang1 activity on TIE2 to foster angiogenesis. Ephrin-B2 links to VEGFR2 or VEGFR3 and promotes their internalization, thereby enhancing angiogenesis. Tip cell selection is associated with inhibition of tip cell formation laterally and migration of tip cells followed by stalk cells during sprout elongation with repression of tip cell fate in stalk cells. Tip and stalk cells can exchange their respective positions during sprouting elongation. The Robo4–Unc5b complex impedes VEGFR signaling. Endothelial cell migration continues in a given direction until anastomosis.
Event | Factors |
|---|---|
Tip cell | VEGFa/c, VEGFR2/3 |
selection | Ang2 |
Inhibition of | DLL4–Notch, |
tip cell fate | Robo4, Wnt, |
in adjacent | VEGFR1 |
endothelial cells | |
Tip cell | Ang1–TIE2 (EC–ECM adhesion sites) |
migration | VEGFa/c/d–VEGFR2, VEGFc/d–VEGFR3/VEGFR2–VEGFR3 |
Sprout | Cadherin-5 extraction from cell junctions |
elongation | MMP (matrix degradation) |
Tip cell | Ephrin-B2–EPHb4, Ephrin-B2–EPHb4–VEGFR2/3, |
guidance | VEGFa/c–Nrp1/2–VEGFR2/3, Sema3e–plexin-D1, |
Slit2–Robo4, Robo4–Unc5b, netrin–Unc5b |
Table 10.2
Endothelial sprouting (Part 2); Source: [886]; ALK: activin receptor-like kinase; Ang: angiopoietin; aPKC: atypical protein kinase-C; EPH: erythropoietin-producing hepatocyte receptor kinase Par: partitioning defective protein; PAK: P21-activated kinase; PDGF: platelet-derived growth factor; PDGFR: PDGF receptor; RasIP: RAS-interacting protein; S1P: sphingosine 1-phosphate; Tβ R: TGFβ receptor; TGF: transforming growth factor; TIE: Tyr kinase with Ig and EGF homology domains (angiopoietin receptor); VEGF: vascular endothelial growth factor; VEGFR: VEGF receptor; vSMC: vascular smooth muscle cell). Prior to tubulogenesis, or lumenogenesis, the apicobasal polarity is established in endothelial cells arranged in a string (precursor vessel). Protein PAR3 is a major determinant of cell polarity that influences lumenogenesis. Cell adhesion proteins (e.g., zonula occludens ZO1, claudin-5, and cadherin-5) move from the apical (wetted) surface to the basolateral segment of the plasma membrane; at the basal surface, integrins connect to matrix constituents. Integrin, PAR3, and RasIP1 promote the lateral redistribution of these junctional components. Lumenogenesis is initiated, at least partly, by relocalization of sialomucin CD34 and podocalyxin (Podxl) to the apical surface mediated by cadherin-5 and β1-integrins. Kinase PKC phosphorylates moesin that links to apical Podxl–CD34 complexes and promotes the deposition of filamentous actin. Podocalyxin may cause an electrostatic repulsion of apical surfaces between endothelial cells. Lumenal expansion may result from vacuole exocytosis and fusion at the apical surface as well as signaling pathways, such as VEGFa–VEGFR2 axis that recruits of myosin-2 to the apical surface and those that activate RoCK to foster actomyosin filament contraction. Certain macrophage populations may act as cellular chaperones for vascular anastomosis.
Event | Factors |
|---|---|
Tubulogenesis | Par3/6, integrins, |
(lumenogenesis) | RasIP1, RhoGAP29, CDC42, Rac1, |
Src, aPKC, PAK2/4, Raf | |
Anastomosis | Macrophage |
Stabilization | PDGFb, TGFβ1 |
and | (PDGFRβ + pericyte and SMC recruitment), |
maturation | S1P–S1P1, Ang1–TIE2, ephrin-B2–EPH |
(mural–endothelial cell attachment), | |
TGFβ1–Tβ R2–ALK5 | |
(vSMC differentiation), | |
deposition of the basement membrane, | |
strengthening of intercellular junctions) |
Endothelial sprouting for angiogenesis requires the coordinated behavior of involved endothelial cells that is regulated by theNotch and VEGFR signalings. Selection of endothelial cells for tip cell position depends on the ratio between VEGFR1 and VEGFR2 receptors [1175]. Receptor VEGFR1 has a high affinity for VEGFa, but a weak kinase activity. It modulates VEGFa signaling via VEGFR2 receptor. Whereas signaling launched by VEGFR2 and VEGFR3 supports tip cells, the decoy receptor VEGFR1 limits tip cell formation [886]. In addition, alternative splicing of VEGFR1 transcript generates a secreted, inactive isoform (soluble VEGFR1S), which serves as a sink for free VEGFa factor.
Alternative splicing of the VEGFA transcript creates many variants with distinct functions. Migration of endothelial cells and, hence, vascular branching are promoted by heparan sulphate-binding VEGFa165 variant. Free VEGFa121 variant influences endothelial cell proliferation, but not migration [886]. Moreover, VEGFR2 output elicited by matrix-bound VEGFa differs from that primed by soluble VEGFa messenger.
Tip cells contains a higher Delta-like (Notch) ligand DLL4 concentration with respect to stalk cells (Table 10.3). Messenger Notch, in turn, determines VEGFR concentration. As afeedback, VEGFR controls DLL4 expression. Endothelial tip cells are activated and guided by an extracellular VEGFa gradient. Activated Notch signaling in stalk cells impedes VEGFR, thereby repressing tip cells and maintaining the hierarchical organization of sprouting tip and stalk cells. The VEGF–VEGFR–DLL4–Notch–VEGFRfeedback loop assigns position of endothelial cells to tip or stalk cells. However, tip cells can shift to stalk cells and conversely according to the feature of VEGFR–DLL4–Notch signaling, which is constantly re-evaluated.
Table 10.3
Mechanisms of endothelial leading, tip and trailing, stalk cell selection and their main features and roles (Source: [886]). Endothelial cells produce multiple Notch receptors (Notch-1, Notch-3, and Notch-4) and transmembrane Notch ligands (DLL1, DLL4, Jag1, and Jag2). Activated VEGFR2 induces DLL4 expression in tip cells that activates Notch on adjacent stalk cells. Ligand binding causes cleavage of Notch receptor by ADAM10, ADAM17, and presenilins, thereby liberating an intracellular fragment (NotchICD). In stalk cells, Notch precludes VEGFR2 activity, downregulates the expression of VEGFR3, and upregulates that of VEGFR1 and soluble VEGFR1, a sink for VEGFa that then represses VEGFR2 activity, thereby preventing tip cell fate. In addition, Notch induces expression of DLL4 and Notch-regulated ankyrin repeat-containing protein (NRARP). The latter supports Wnt signaling in stalk cells, thus maintaining adhesion sites between endothelial cells; it also fosters inhibitory feedback of Notch. In tip cells, Notch is blocked by Jagged-1 produced by stalk cells; it impedes DLL4–Notch connections on tip cells when Notch is glycosylated. Binding of VEGFc to its VEGFR3 receptor, which is highly expressed in tip cells, enables lymphangiogenesis.
Tip cell | Stalk cell |
|---|---|
VEGF signaling | DLL4–Notch signaling |
VEGFa/c | Low VEGFR signaling |
VEGFR2/3 | |
Low Notch signaling | |
PDGFb, Unc5b | |
Highly motile | Weakly motile |
Guide sprouting | Lumenogenesis |
Maintenance of between-cell junctions | |
Connection to parent vessel |
Endothelial sprouts are converted into functional vessels. Sprout extension involves the local proliferation and migration of the endothelial cells behind the tip that forms the sprout stalk. Tip cells do not proliferate.
Navigation cues are sensed by tip cells. The navigators Uncoordinated-5 homolog Unc5b, Roundabout homolog Robo4, plexin-D1, neuropilins, ephrin-B2, and EPHb4 receptor are major conductors of angiogenesis. The growing endothelial sprout is guided by attractive (e.g., netrins) and repulsive cues (e.g., semaphorin-3). MicroRNAs ensure the post-transcriptional control of angiogenesis.
Establishment of blood flow requires the formation of a vascular lumen. Cellular structures are converted into tubes by vacuole formation and intracellular and subsequent intercellular fusion of large vacuoles. Vascular tubulogenesis is initiated by the acquisition of the apicobasal polarity of endothelial cells that is regulated by cell–matrix interactions and signaling via partitioning defective protein PAR3 and VEGFR receptor. Tubulogenesis is controlled by EGF-like domain-containing protein EGFL7 expressed by endothelial cells.
Interactions between tip cells regulate the fusion of adjoining sprouts to form a continous lumen. Tip cells abandon exploration upon encountering tips of other sprouts or existing capillaries, i.e., when they can form new vascular connections. Junctional contacts are then built at merging regions. Newly formed vessels are stabilized by recruitment of pericytes and deposition of matrix proteins into a basement membrane.
10.2.4 Other Modes of Vessel Formation and Remodeling
Changes in the local balance between pro- and anti-angiogenic factors can lead to the elimination of new connections (pruning). The splitting of vessels through the insertion of tissue pillars (intussusception) expends the vascular network.
Intussusception (“growth within itself”), a non-sprouting angiogenesis, represents a particular way of expanding and modifying a vessel network, as it creates and remodels blood vessels via the formation of transluminal pillars [1176].6 Regions of locally changed stress sensed by endothelial cells can trigger growth of septa, ridges, pillars, and folds. Intussusception predominantly occurs in regions with accelerated blood flow. Intussusceptive vascular growth and remodeling are regulated by vascular growth factors, such as VEGF, PDGFb, and angiopoietins, and TIE receptors.
Maturation is related to the transition from a growing vascular bed to a functional network, characterized by stabilized vessels with mural cells and a basement membrane, valved veins, capillaries, and lymphatics with either fenestrations or tight junctions.
Perfusion reduces hypoxia-induced angiogenic factors and promotes vessel maturation. Bone marrow-derived circulating cells can be retained in the perivascular space due to CXCL12 chemokine in response to VEGFa and then enhance endothelial proliferation.
10.2.5 Lumenogenesis
The apical membrane initiation site (AMIS) corresponds to one of the earliest intermediates in lumenogenesis [1177]. This cell-surface compartment as well as subjacent vesicles are characterized by the transient accumulation of numerous apical polarity and transfer proteins. The delivery of apical cargos to specialized zones of intercellular contacts, AMISs, i.e., the forming lumen, requires exocytosis.7
The apical membrane initiation site generates the preapical patch, a closed lumen formed by the newly established apical membranes of adjacent cells that is bound at its margins by junctional complexes. Opening of the lumen is a result of ion and water transport. Further organization of membranes, junctional complexes, cytoskeleton, and organelles creates the mature cyst, i.e., the tight junction-delineated lumen.
Lumenogenesis thus depends onRab11a, Rab8a, and Rab8b GTPases andguanine nucleotide-exchange factor Rab8-interacting protein (RabIn8) [1178].8 Lumen formation, at least in nephron epithelial cells, relies on a Rab11a–RabIn8–Rab8a axis that recruits Sec15a and cortical polarity GTPaseCDC429 and promotes apical exocytosis by enrolling thepartitioning-defective protein (Par) complex with members Par3 and atypical protein kinase-C and multimericexocyst-tethering complex subunit Sec10, and exocyst subunit Sec8 to small vesicles and multiple rudimentary lumens and close to sites of intercellular contacts that constitute early apical membrane initiation loci.
Rab11a + recycling endosomes are involved in the transfer of apical proteins.10 Exocytosis that depends on Rab11a involvesRab8GEF RabIn8; the latter recruits and activates Rab8 GTPase.11 Activation of Rab8 is antagonized by Rab8GAPTBC1D30 (Vol. 4 –Chap. 9. Guanosine Triphosphatases and Their Regulators).12 Small Rab11a GTPase and possibly Rab8 recruit the exocyst subunit Sec15a that promotes the binding of the exocytic carrier to the Sec10 exocyst subunit on the emerging AMIS membrane. Exocytosis also permits the initial recruitment of the Par3–aPKC complex to AMIS sites. Active Rab8 stimulates CDC42 interaction with the exocytic carriers, probably via CDC42GEF DnmBP or Tuba.13 Small GTPase CDC42 recruits atypical protein kinase-C that further promotes AMIS formation and exocytosis, as it recruits exocyst protein Sec10 and Sec8 as well as Par3 that colocalizes with Sec8 protein.
10.3 Arteriogenesis
Once the lumen of a main artery is strongly narrowed, the lumen of small arteries increases to form collaterals that can maintain the blood perfusion. Arteriogenesis is defined as the remodeling of a part of a pre-existing arterial network outside anischemic region to form functional irrigation arteries.
Arteriogenesis is initiated by CCL2 chemokine. Chemoattractant CCL2 stimulates the formation of a collateral circulation on arterial occlusion [1181]. Arteriogenesis thus involves inflammation with monocyte recruitment.
Various substances are also required at different stages of arteriogenesis, among these, TGFβ, PDGF, FGF2, CSF2, and TNFα. Attracted monocytes produce fibronectin and proteoglycans as well as peptidases to remodel the extracellular matrix. These inflammatory cells then produce growth factors to stimulate proliferation ofendothelial andsmooth muscle cells.
10.4 Vasculature Compartments
Hemangioblasts give birth to angioblasts that generate vascular endothelial cells. During vasculogenesis, endothelial precursor cells in response to local signals undergo proliferation, migration, differentiation, specification, and coalescence to form the lining of nascent vessels. During angiogenesis, the vascular network is remodeled into arteries, veins, and capillaries.
10.4.1 Lumen Size and Vessel Architecture
Whether blood flows with high or low flow rates and/or rapidly or slowly, vessels widen or narrow, respectively. Blood vessels that carry a high-speed, large-volume flow enlarge, whereas those with low-speed, small-volume flow regress. Local features of hemodynamics thus direct formation of main perfusion vessels, as endothelial cells respond to hemodynamical forces. Therefore, mechanical forces generated by blood flow modify gene expression in the developing embryo. Blood flow contributes to shaping a functional vascular architecture although vessel identity and developmental patterning are genetically predetermined.
The vessel network is organized according to neighboring tissue growth and structure. Vessel positioning depends on traction and compression forces exerted by the growing tissues.
10.4.2 Cost Functions
Optimal design of vessel branching is based on cost functions that are the sum of the rate at which work is done on blood and the rate at which energy is used, which are supposed to be proportional to the vessel volume for each vessel segment [1182]. Other cost functions have been proposed based on the minimal total surface area of blood vessels, minimal total volume, or the minimal total wall shear force on the vessel wall, or minimal power of the blood flow.
10.4.3 Arteriovenous Differentiation
The primitive vascular network progressively matures with arteries, capillaries, and veins [1183]. The specification of vascular cells in the different compartments (arteries, capillaries, and veins) of the vascular circuit is not only determined by applied mechanical forces and chemical factors (activators, inhibitors, and hypoxia), but it is also genetically programmed. Specific markers assigned to cells in each compartment can be detected before the onset of circulation [1167].
Growth and specialization of arteries and veins continues throughout development. Blood vessels adapt to experienced hemodynamic stresses. Arteriovenous differentiation is controlled by hemodynamic factors. Blood vessels remodel according to loading history. Concentric layers of vascular smooth myocytes and elastic fibers ensure resistance to arteries. The venous low-pressure system is provided with valves that open unidirectionally to prevent backflow.
Vasculature development depends on a combination of intrinsic prepatterning and extrinsic responses to environmental parameters. The direction of moving blood determines the differentiation in artery or vein, whether the lateral branch receives blood or provides it. Arterial endothelial cells characterized by a spindle-like shape in the streamwise direction have a low proliferation rate.
Some genes control the vascular specification into arteries and veins via signaling molecules (Table 10.4). Arterial endothelial cells express certain transcription factors, signaling molecules, gap-junction proteins, matrix molecules, and adhesion proteins that are absent from veins.
Table 10.4
Molecules involved in arterial and venous differentiation of endothelial cells. Notch targets 2 families of transcriptional repressors: hairy and enhancer of split (HES) and HES-related (HRT) transcriptional regulators. Sonic Hedgehog and VEGF induce arterial cell fate. Forkhead box-C transcription factors FoxC1 and FoxC2 control arterial specification via Delta-like ligand DLL4, a Notch agonist. Nuclear receptor NR2f2 (a.k.a. chicken ovalbumin upstream promoter transcription factor COUPTF2) suppresses the Notch pathway and inhibits neuropilin-1 expression. Ephrin-B2 and ephrin receptor EPHb4 are markers for arteries and veins, respectively. Phosphatidylinositol 3-kinase inhibits the phospholipase-Cγ1–PKC–MAP2K–ERK pathway, an effector of VEGF signaling for arterial fate. Protein kinase-B induces venous fate.
Arterial fate | Venous fate |
|---|---|
VEGFa | VEGFR3 |
Neuropilin-1 | Neuropilin-2 |
Notch | NR2f2 |
Ephrin-B2 | EPHb4 |
Extracellular signal-regulated kinase | Phosphatidylinositol 3-kinase |
The arteriovenous differentiation is done via different processes.Notch guides arterial fate, as it targetsHairy and enhancer of Split (HES) andHES-related (HRT) transcriptional regulators. In arteries, Notch signaling is indirectly stimulated by VEGFa via VEGFR2 orneuropilin-1 and promoted byforkhead box transcription factors FoxC1 and FoxC2 [1184]. One the other hand, Notch pathway in the venous endothelium is suppressed by NR2f2 nuclear receptor.14
Arterial expression profile is defined by markerephrin-B2, whereas venous pattern is identified by markerEPHb4 receptor. Neuropilin-1 is found in arteries, whereas Nrp2 is restricted to veins and lymphatic vessels.
10.5 Extracellular Matrix in Vessel Formation
Angiogenesis depends on many growth factors and enzymes, hence different cell types and surrounding medium. The behavior of endothelial cells, especially their migration and proliferation as well as formation of tubular structures is influenced by theextracellular matrix (Table 10.5). Tip cells produce peptidases for cell migration, such as membrane-typematrix metallopeptidase mt1MMP (or MMP14). Nitric oxide, prostaglandin-E2, and CCL2 chemokine increase the cell-surface clustering and activity of mt1MMP [1185].
Table 10.5
Sprout endothelial cells and inflammatory and angiogenic agents (Source: [1185]; VEGFR1S: soluble VEGFR1). A nascent vascular sprout contains 3 types of endothelial cells. Tip cells produce VEGFR2, VEGFR3, DLL4, and angiomotin, among others, and navigate into the surrounding tissue, but proliferate poorly. Stalk and phalanx cells form the lumen and promote nascent vessel stabilization.
Type | Stimulators |
|---|---|
Tip | VEGF, S1P, |
Bdk, TNFα | |
Stalk | EGFL7, FGF, VEGF, VEGFR1S |
Phalanx | Ang2, BMP9, FGF2, VEGF, |
Tsp |
Interactions between endothelial and surrounding cells, such as pericytes and vascular smooth myocytes, regulate not only vessel stabilization and remodeling, but also vascular formation. Intercellular communications are based on multiple molecules, such as transforming growth factor-β, angiopoietins, platelet-derived growth factor, sphingosine 1-phosphate, and Notch ligands, among others [1186].
Cell shape is coupled to proliferation. Cell division depends more on the degree of possible extension than the level of matrix binding. The actin cytoskeleton state and activity of myosin contribute to cell and tissue growth. Laminin, fibronectin, andcollagen-1, -3, -4, and -5 promote cell spreading.
Rho GTPases are involved in tension-dependent growth control, because they regulate cytoskeletal contractility. Matrix-associated cytoskeletal mechanics can explain tension-driven tissue modeling. Podosomes of endothelial cells, sites of MMP concentration, are involved in endothelial cell migration and angiogenesis.
10.5.1 Growth Factors
Many growth factors such as VEGF bind to matrix constituents.
10.5.1.1 VEGF
Vascular endothelial growth factors connected to matrix constituents can be cleaved. A C-terminus of variable length according to the involved peptidase is then released. Released VEGF from matrix stores promotes angiogenesis.
Various VEGF isoforms have different affinities for given matrix proteins. However, soluble VEGF subtypes (VEGFS) lack the matrix-binding region. They are either secreted as short alternative spliced forms such as VEGF120 or cleaved by plasmin ormatrix metallopeptidases, such as MMP3, MMP7, MMP9 (in the presence of heparin), and MMP19 isozymes.
Matrix-bound and soluble VEGFs can both trigger VEGFR phosphorylation, but differ in signaling. Matrix anchorage leads to clustering of VEGFR2, promotes receptor endocytosis, and raises downstream phosphorylation kinetics. Soluble VEGF has a tendency to build low-density, poorly branched vascular networks (vascular hyperplasia) [1185]. On the other hand, matrix-bound VEGF favors highly branched vessels.
10.5.1.2 FGF
Fibroblast growth factors interact with matrix proteins, particularly heparan sulfate proteoglycans. The extracellular matrix contributes to the regulation of FGF signaling. In pancreatic β cells, FGFR1 concentration is reduced by the binding ofα6-integrin to laminin [1185].
10.5.1.3 PDGF
Platelet-derived growth factor can link to several collagen types, laminin-1, nidogen, and perlecan. Binding to matrix constituents may serve for storage in cell’s surrounding for later release and signaling.
10.5.1.4 TGF
Transforming growth factor-β can anchor to the extracellular matrix. This anchorage can contribute to its activation. Thrombospodin binding stimulates TGFβ, but thrombospodin-1 inhibits VEGF factor.
10.5.2 Protein Fragments and Peptidases of the Extracellular Matrix
Proteic constituents and peptidases of the extracellular matrix contribute to angiogenesis. Released active fragments of cleaved matrix constituents, the so-calledmatrikines, are involved in the control of angiogenesis and healing (Table 10.6). Most of the matrikines compete with intact matrix components for interaction withintegrins.
Table 10.6
Examples of matrikines in inflammation and angiogenesis (Source: [1185]; I: integrin; SPARC: secreted protein acidic and rich in cysteine; Tsp: thrombospondin). Matrikines are fragments of matrix constituents generated by proteolytic cleavage with functions distinct from those of parental proteins.
Matrikine | Source | Receptor | Function |
|---|---|---|---|
Anastellin | Fibronectin | Anti-angiogenic | |
Angiostatin | Plasminogen | αVβ3, α9β1-I | Anti-angiogenic |
Arresten | Collagen-4 | α1β1-I | Anti-angiogenic |
Canstatin | Collagen-4 | Anti-angiogenic | |
Endorepellin | Perlecan | Anti-angiogenic | |
Endostatin | Collagen-18 | αVβ3-I | Anti-angiogenic |
Metastatin | Collagen-4 | Anti-angiogenic | |
Neostatin | Collagen-18 | Anti-angiogenic | |
Tumstatin | Collagen-4 | αVβ3, α5β1-I | Anti-angiogenic |
Vastatin | Collagen-8 | Anti-angiogenic | |
Restin | Collagen-15 | Anti-angiogenic | |
Peptides from | Elastin | Pro-angiogenic | |
Peptides from | SPARC | Pro/anti-angiogenic | |
Peptides from | Tsp | Pro/anti-angiogenic | |
Peptides from | Collagens-1/4, | Elastin-binding protein, | Chemotactic |
elastin, | L-selectin, | ||
fibronectin, | integrins, | ||
laminins, | CXCR1/2 | ||
entactin, Tsp, | |||
hyaluronan |
Many matrikines have an anti-angiogenic effect. They derive from components of: (1) the basement membrane, such as collagen-4,15 perlecan,16 and fibronectin,17 as well as (2) the interstitial matrix that surrounds the vasculature, such as collagen-18,18 collagen-8,19 and collagen-15.20
Both thrombospondin-1 and -2 as well as their proteolytically derived fragments hinder angiogenesis during wound healing. Thrombospondin fragments are more effective than full-length thrombospondins.
In addition, peptidic fragments that are cleavage products of collagen-1 and -4, elastin, fibronectin, laminins, nidogens, thrombospondins, and hyaluronan operate as chemotactic substances for inflammatory cells (Table 10.7).
Peptidic fragments | Effect |
|---|---|
Collagen | Neutrophil recruitment |
Elastin | Monocyte recruitment |
Laminin | Cell migration |
These fragments can also induce specific gene expression programs inleukocytes, in particular those of peptidases and cytokines (Table 10.8). In particular, fragments of fibronectin and laminin increase the production of various peptidases such asmt1MMP in monocytes and macrophages [1185].
Table 10.8
Peptides released from cleavage of matrix constituents and production of peptidases and cytokines (Source: [1185]; MMP: matrix metallopeptidase; PAI: plasminogen activator inhibitor; uPA: urokinase-type plasminogen activator [urokinase]).
Peptidic fragments | Effect |
|---|---|
Collagen | Production of peptidases and cytokines |
Elastin | Production of peptidases and cytokines |
Fibronectin | Production of MMP9/12, cytokines |
Hyaluronan | Production of MMP12, PAI1, cytokines |
Laminin-111 | Production of MMP9, uPA |
Laminin-511 | Production of MMP9/14, cytokines |
Secreted protein acidic and rich in cysteine (SPARC)21 is a member of the group of non-structural components of the extracellular matrix. This secreted Ca2 + -binding glycoprotein modulates cell adhesion and proliferation, as it intervenes in interactions between cells and their environment [1187].22 It is highly expressed in various cell types in remodeling tissues. It can interfere with the binding of angiogenic stimulators, such as vascular endothelial (VEGF), platelet-derived (PDGF), and fibroblast (FGF2) growth factor, to their receptors in endothelial cells. Schwann cells influence neuroblastoma growth by secreting inhibitors of angiogenesis, the most potent of which is SPARC protein. Full-length SPARC (SPARCFL) and SPARC-derived peptides (SPARCf) that correspond to the follistatin domain of the protein as well as its N- and C-termini block angiogenesis [1189].23
Involved peptidases includematrix metallopeptidases mt1MMP (MMP14) to mt3MMP (MMP16), MMP2, MMP3, MMP7, MMP9, and MMP13, and members of the adamlysine family, such asADAM10, ADAM15, ADAM17, and ADAMTS1 [1185].
10.5.2.1 Endostatin and Endorepellin – Endogenous Angiogenesis Inhibitors
Endostatin, a fragment of collagen-18, andendorepellin, an inhibitor of angiogenesis derived from the basement membrane proteoglycanperlecan, have dual roles as structural constituents and functional regulators of tissue growth. They modulate the activity of growth factors. They can inhibit the growth of blood vessels and stabilize the basement membrane [1190].
Endostatin increases the production ofnitric oxide in endothelial and smooth muscle cells [1191]. Hypertension is a major drawback of VEGF inhibitor administration, which necessitate to limit the dose. On the other hand, NO, a potent vasodilator, lowers blood pressure. Endostatin reduces blood pressure via the NOS3–NO axis and/or improved NO availability.
10.5.3 Cell Adhesion Molecules
Angiogenesis is regulated by endothelial cell adhesion molecules, such as vascular endothelial cadherin, integrins, and platelet–endothelial cell adhesion molecule. A mechanosensory complex composed of PECAM, cadherin-5, and VEGFR2 causes the response initiated by activated integrins [1192].
α-(2,6)-Sialic acid is necessary for the cell-surface residency and maintenance as well as homophilic interactions of platelet–endothelial cell-adhesion molecules [1192]. This cell adhesion molecule is involved in angiogenesis once PECAMs are linked, and also in mechanical stress sensing and its anti-apoptotic function.
10.5.3.1 Integrins
Integrins are implicated as mediators of vascular formation and homeostasis, due to their role in cell adhesion, migration, positioning, proliferation, and differentiation [1193]. Endothelial cells express different vascular integrins involved in vasculo- and angiogenesis such as αVβ3-integrins.
Plasmalemmal integrins that interact with the extracellular matrix promote cell attachment to the extracellular matrix and transduce signals to the nucleus. Integrin-mediated adhesion to the extracellular matrix activates theERK pathway. Activated extracellular signal-regulated kinases stimulate cyclin-D1. Cell division is controlled by cyclins, cyclin-dependent kinases, and CDK-inhibitors [1194] (Vol. 2–Chap. 2. Cell Growth and Proliferation).
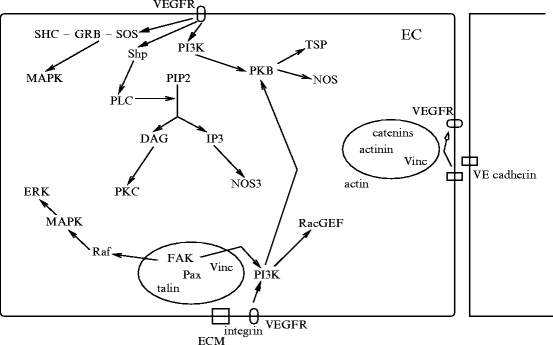
Integrins are also required in VEGF signaling viaRas GTPase orphosphatidylinositol 3-kinase. Integrin-binding protein lactadherin 24 is involved in VEGF-dependentneovascularization [1195]. Lactadherin interacts with 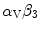 – and
– and 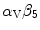 -integrins and alters VEGF-dependent PKB phosphorylation and neovascularization.
-integrins and alters VEGF-dependent PKB phosphorylation and neovascularization.
– and -integrins and alters VEGF-dependent PKB phosphorylation and neovascularization.Activated 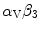 -integrin supports angiogenesis and growth of brain metastasis via upregulation of vascular endothelial growth factor under normoxic conditions [1196]. Synthesis of VEGF is post-transcriptionally controlled by phosphorylation (inhibition) of translational repressor 4E-binding 4eBP1 protein.
-integrin supports angiogenesis and growth of brain metastasis via upregulation of vascular endothelial growth factor under normoxic conditions [1196]. Synthesis of VEGF is post-transcriptionally controlled by phosphorylation (inhibition) of translational repressor 4E-binding 4eBP1 protein.
-integrin supports angiogenesis and growth of brain metastasis via upregulation of vascular endothelial growth factor under normoxic conditions [1196]. Synthesis of VEGF is post-transcriptionally controlled by phosphorylation (inhibition) of translational repressor 4E-binding 4eBP1 protein.10.5.3.2 Tenascins
The tenascin (Ten) family of matrix, cell adhesion glycoproteins (Table 10.9) contributes to vasculogenesis and various normal and pathological processes of mature life, such as wound healing and vascular diseases [1197].
Table 10.9
Family of tenascins (Ten). These matrix, cell adhesion glycoproteins act via integrins, cell adhesion molecules of the immunoglobulin superfamily (IgCAMs), protein Tyr phosphatase receptor PTPRz1, annexin-2, fibronectin, and matrix proteoglycan lectican. The tenascin family includes TenC, TenR, TenW, TenX, and TenY. Tenascins can have anti-adhesive effects. Tenascin-C can promote integrin-dependent protein clustering at focal adhesions, as it interacts with RTKs and activates ERK. Like TenC, TenX (or flexilin) is expressed in the heart as well as vascular smooth myocytes. TenY is coexpressed with TenC in the lungs.
Tenascin | Alias |
|---|---|
TenC | Cytotactin |
TenR | Restrictin, janusin |
TenW | |
TenX | Flexilin, hexabrachion |
TenY |
In association with matrix proteins and plasmalemmal receptors such as integrins, tenascins have opposite cellular functions, according to the mode of presentation, cell type, and differentiation state of the target tissues. Tenascins are regulated by growth factors, vasoactive peptides, matrix proteins, and mechanical factors [1197].
Tenascin-C interacts with integrins, collagens, proteoglycans, and fibronectin. It behaves either as an adhesive or anti-adhesive protein. It binds to annexin-2, a plasmalemmal receptor of endothelial cells [1198]. The TENC gene expression is mechanosensitive.
10.5.4 Fibulins
Fibulins (Fbln1–Fbln7; Table 10.10) constitute a family of secreted calcium-binding glycoproteins that are encoded by 7 genes. They interact with several matrix proteins, such as fibrillin, fibronectin, laminins, proteoglycans, and tropoelastin.
Table 10.10
Family of fibulins (Fbln [Fibl]; ARMD: age-related macular degeneration; AxPC1: ataxia, posterior column 1, with retinitis pigmentosa; DANCE: developmental arteries and neural crest EGF-like protein; DHRD: Doyne honeycomb retinal dystrophy (macular degeneration) protein; EFEMP: EGF domain-containing fibulin-like extracellular matrix protein). Fibulins regulate organ shape with growth factors and stromal cells. They are involved in cell proliferation, migration, differentiation, and survival.
Type | Other aliases |
|---|---|
Fbln1 | Fibl1 |
Fbln2 | Fibl2 |
Fbln3 | Fibl3, EFEMP1, DHRD |
Fbln4 | Fibl4, EFEMP2 |
Fbln5 | Fibl5, DANCE |
Fbln6 | Fibl6, hemicentin-1 (Hmcn1), ARMD1, AXPC1 |
Fbln7 | Fibl7 |
Fibulin-1 is associated with elastic fibers. It lodges particularly in basement membranes. It is also present in blood, where it binds to fibrinogen. It is incorporated in fibrin clots. Fibulin-1 suppresses fibronectin-mediated adhesion and motility [1199].
Fibulin-2 is involved in microfibril and elastic fiber organization [1200]. It can participate in clusters made of nidogen-1 and -2, fibronectin, and perlecan, a major heparan sulfate proteoglycan in the basement membrane that possesses immunoglobulin-like modules (IG2-IG15) with many binding sites for cellular and extracellular ligands [1201].
Fibulin-3 stimulates production of vascular endothelial growth factor, hence angiogenesis [1202]. Moreover, fibulin-3 hinderstumor cell apoptosis.
Fibulin-4 is an elastic fiber-associated protein that is required for formation of elastic fibers, especially in arteries, lungs, and skin. It regulates tropoelastin expression [1203].
Fibulin-5 that interacts with integrins is expressed in developing arteries. Its expression is reinduced in injured vessels and atherosclerosis in intimal smooth muscle and endothelial cells [1204].
10.5.5 Thrombospondins
Vessel formation requires a tuned balance between various interacting stimulatory and inhibitory signals.Thrombospondin inhibits blood vessel formation. Thrombospondin-1 binds to integrins,matrix metallopeptidase-2, transforming growth factor TGFβ1, proteoglycans, and fibronectin.
10.5.6 Matrix Metallopeptidases
Remodeling of the extracellular matrix by various molecules, especially extracellular and membrane-boundmatrix metallopeptidases, is required during angiogenesis. The type of cleavage of matrix components can lead to opposite effects. Proteolysis of collagen-4 and -18, as well as plasminogen, releases angiogenesis inhibitors, whereas cleavage of collagen-4 by MMP9 stimulates migration of endothelial cells and angiogenesis.
10.5.7 Transglutaminase and Carboxypeptidases
Angiogenesis requires localized destabilization of the extracellular matrix. Afterward,transglutaminase-2 is downregulated in endothelial cells during capillary morphogenesis to secondarily stabilize the underlying basement membrane [923].Glutamate carboxypeptidase-225 expressed in endothelial cells, contributes to the remodeling of the extracellular matrix for angiogenesis. Glutamate carboxypeptidase-2 acts on β1-integrin and leads to activation of P21-activated kinase PAK1 [1205]. It also interacts with actin-binding filamin-A, leading to phosphorylation of P21-activated kinase-1.26 Glutamate carboxypeptidase-2 thus participates in an autoregulatory loop.
10.5.8 Collagen Prolyl 4-Hydroxylase
Transcription factor P53 activates the gene that encodes collagen prolyl 4-hydroxylase. The latter is required for extracellular release of collagen-derived peptides, such as endostatin and tumstatin that inhibit angiogenesis [1206].
10.6 Mediators of Vascular Formation
The formation of new blood vessels, the patterning of the ramified network,27 as well as the remodeling of existing blood vessels require the migration and proliferation of various cell types controlled by manifold substances, such as guidance and growth factors, and their inhibitors (Table 10.11). Microvascular growth and remodeling, which is influenced by the mechanical factors (local stress field), are variable. These phenomena are indeed based on sprouting, non-sprouting (intussusceptive), and mixed angiogenesis.
Table 10.11
Angiogenesis receptors and their ligands. Crosstalks between signaling effectors allow to coordinate their spatial and temporal activities.
Receptor | Ligand |
|---|---|
VEGFR | VEGF (endothelial cell) |
Neuropilin | Semaphorin |
PDGFR | PDGF (smooth myocyte) |
HER | EGF |
FGFR | FGF |
TGFR | TGF |
TNFR | TNF |
EPH | Ephrin |
TIE | Angiopoietin |
S1PR | S1P |
Roundabout | Slit |
Netrin | Uncoordinated-5 |
Galectin-1 | Anginex |
10.6.1 Navigation Signals
Angiogenesis is conducted by an initiation and patterning program. Guidance molecules regulate patterning of blood vasculature. The navigation of migrating angiogenic components requires guidance signaling via ligand–receptor complexes, such as: (1) ephrins withEPH receptors; (2) Slit ligands withRoundabout (Robo) receptors; (3) semaphorins withplexin andneuropilin receptors; and (4) netrins with plasmalemmal receptorsDeleted in colorectal carcinoma (DCC), DCC-relatedneogenin (Ngn), anduncoordinated-5 (Unc5) receptors [1207, 1208].28
Nerve and vessel navigations to targets are subjected to a cooperative procedure, axonic and angiogenic signals guiding both vessels and axons. Ephrins, semaphorins, netrins, and slits29 control vessel morphogenesis.
10.6.1.1 Semaphorins
10.6.1.2 Semaphorin-3 Subtypes
During blood vessel genesis, endothelial cells producesemaphorin-3 that controlsintegrin function for vascular reshaping [1209]. Semaphorin-3A expressed by endothelial cells of developing vessels inhibits endothelial cell migration at nascent adhesive sites of spreading endothelial cells, by interacting with integrins, such as endostatin and thrombospondin.
Neuropilin-1 is a membrane receptor for multiple ligands30 such as semaphorin-3 that regulates developmental process guidance [1210, 1211].
Like Sema3a, Sema3f influences vessel formation via neuropilin-1. Dimer Sema3e that targetsplexin-D1 is a chemorepellant of endothelial cells [1212]. Agent Sema3e controls endothelial cell positioning and patterning of the developing vasculature. One the other hand, catabolized Sema3e can act as a chemoattractant.
10.6.1.3 Semaphorin-4 Isoforms
Semaphorin-4A, an activator of T-cell-mediated immunity, is also expressed in endothelial cells. Semaphorin-4A hinders endothelial cell migration and division stimulated by vascular endothelial growth factor, thus preventing angiogenesis in vivo [1213]. Semaphorin-4A uses different receptors according to the cell type. It targets plexin-D1 in endothelial cells with a lower affinity for plexin-D1 than Sema3e.
As an activator of T-cell-mediated immunity, Sema4a connects to T-cell immunoglobulin and mucin domain-containing protein TIMD2 in T lymphocytes. Semaphorin-4A also binds to Plexin-B1. It uses PI3K effectors in the VEGF–VEGFR2 pathway. It suppresses PKB phosphorylation and production of RacGTP that promotes endothelial cell motility.
Semaphorin-4D induces endothelial cell proliferation and tubule formation via plexin-B1 [1214]. Plexin-B1 activates HGFR receptor protein Tyr kinase.
10.6.1.4 Netrins
Netrins (sanskrit netr: scout) are secreted matrix-binding proteins that yield migration signals for axon guidance and motion of other cells in the developing central nervous system. Axon-guidance functions are mediated by 2 families of receptors: (1) Uncoordinated-5 (Unc5) receptor family and (2) Deleted in colorectal cancer (DCC) family that includes neogenin subclass and belongs to the immunoglobulin superfamily. Netrins provide attractive or repulsive signals by interacting with DCC and Unc5 receptors or Unc5–DCC heterodimers, respectively.
Calcium influx via plasmalemmal Ca2 + channels and from cytoplasmic stores is required. High and low cAMP-to-cGMP ratios favor attraction and repulsion, respectively. Netrins also regulate cell adhesion, motility, proliferation, differentiation, and survival in other biological tissues.31
Netrins are guidance cues for vascular endothelial and smooth muscle cells during angiogenesis. Netrin receptor Unc5b in endothelial tip cells and developing capillaries mediates repulsion.Integrin-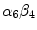 and –
and –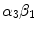 operate as netrin-1 receptors in endothelial cells [1215]. Activated integrins recruit and phosphorylate adaptorSHC that, in association with theGRB–SOS complex, activatesRas, thenRaf andMAPK module. Adaptor SHC also activatesphosphatidylinositol 3-kinase andprotein kinase-B.
operate as netrin-1 receptors in endothelial cells [1215]. Activated integrins recruit and phosphorylate adaptorSHC that, in association with theGRB–SOS complex, activatesRas, thenRaf andMAPK module. Adaptor SHC also activatesphosphatidylinositol 3-kinase andprotein kinase-B.
and – operate as netrin-1 receptors in endothelial cells [1215]. Activated integrins recruit and phosphorylate adaptorSHC that, in association with theGRB–SOS complex, activatesRas, thenRaf andMAPK module. Adaptor SHC also activatesphosphatidylinositol 3-kinase andprotein kinase-B.Netrins activate small GTPasesRac,Rho, andCDC42 (Table 10.12). Netrin-1 bound to DCC recruitsextracellular signal-regulated kinases ERK1 and ERK2 to a DCC receptor complex. Receptor DCC also interacts withfocal adhesion kinases andSrc kinases.
GTPases | CDC42, Rac, Rho |
Kinases | FAK, MAPK, PAK, PI3K, PKA, PKG, PLCγ, Src |
Transcription factors | ELk1, NFAT, P53 |
Netrin-1 acts a survival cue. Once bound to DCC and Unc5h, it activates caspase-3 that cleaves the death domains of these receptors, the latter activating apoptotic caspase-9. Moreover, Unc5b targeted by P53 mediates P53-dependent apoptosis until it binds netrin-1.
10.6.1.5 Ephrins
Ephrin-A1 mediates TNFα-induced angiogenesis in vivo [1216]. Ephrin-B2 and its receptor EPHb are arterial markers. Receptor EPHb activatesSrc kinase via a cleavage of ephrin-B2, releasing a cytoplasmic C-terminal fragment (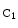 ephrin-B2) that is further processed by thepresenilin-1–γ-secretase complex to produce intracellular
ephrin-B2) that is further processed by thepresenilin-1–γ-secretase complex to produce intracellular 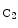 ephrin-B2 peptide.32 Peptide
ephrin-B2 peptide.32 Peptide 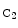 ephrin-B2 binds Src kinase, thereby impeding Src association withCSK kinase, a Src inhibitor, and allowing Src autophosphorylation [1217].33 Activated Src phosphorylates ephrin-B2, inhibiting its processing by γ-secretase and triggering the recruitment ofGRB4 to ephrin-B2. Adaptor GRB4 controls actin dynamics and cell migration, thereby sprouting of endothelial cells.
ephrin-B2 binds Src kinase, thereby impeding Src association withCSK kinase, a Src inhibitor, and allowing Src autophosphorylation [1217].33 Activated Src phosphorylates ephrin-B2, inhibiting its processing by γ-secretase and triggering the recruitment ofGRB4 to ephrin-B2. Adaptor GRB4 controls actin dynamics and cell migration, thereby sprouting of endothelial cells.
ephrin-B2) that is further processed by thepresenilin-1–γ-secretase complex to produce intracellular ephrin-B2 peptide.32 Peptide ephrin-B2 binds Src kinase, thereby impeding Src association withCSK kinase, a Src inhibitor, and allowing Src autophosphorylation [1217].33 Activated Src phosphorylates ephrin-B2, inhibiting its processing by γ-secretase and triggering the recruitment ofGRB4 to ephrin-B2. Adaptor GRB4 controls actin dynamics and cell migration, thereby sprouting of endothelial cells.Endothelial cells express guidance molecules for angiogenesis, such as EPHb4 and its ligand ephrin-B2 [1218]. EPHb4 is a negative regulator of blood vessel branching, leading to circumferential vessel growth rather than angiogenic sprouting and vessel interconnection. EPHb4 and ephrin-B2 restrict migration of endothelial cells. Moreover, EPHb4 reduces vascular permeability viaangiopoietin-1–TIE2 activation at the endothelium–pericyte interface. EPHb4 reverse signaling via ephrin-B2 represents the predominant signaling pathway, being independent of EPHb4 RTK activity and EPHb4 forward signaling.
10.6.1.6 Slit Ligands and Roundabout Receptors
Slit ligands (Slit1–Slit3) are involved in neuronal and vascular development. They bind toRoundabout receptors (Robo1–Robo4) that belong to the immunoglobulin superfamily of transmembrane signaling molecules. The Slit–Robo signaling induces repulsion. Slits are involved in heart morphogenesis, angiogenesis, and tumormetastasis.
Vascular endothelium-specific Roundabout-4 has an extracellular domain different from that of all other Robo family members. Receptor Robo4 activated by Slit2 maintains vasculature integrity [892]. Stabilizing factor Robo4 is expressed in developing blood vessels where it can neutralize VEGF, but it is not required for developmental angiogenesis. During sprouting, Robo4 is only expressed by more mature, stabilized endothelial cells that form sprout stems. The Robo4–Slit2 signaling counteracts hyperpermeability induced by VEGF that uses Src and Yes kinases. In addition, activated Robo4 impedes endothelial cell migration primed by fibroblast growth factor-2 [1219]. Angiogenic endothelial cells (as well as many other cell types) also express Robo1 receptor. Because Slit2 binding to Robo1 promotes angiogenesis, Slit2 positively and negatively regulates angiogenesis by binding to Robo1 and Robo4, respectively.
10.6.2 Transcriptional Regulators
10.6.2.1 Hairy and Enhancer of Split-Related Transcription Factors
Two genes — Hrt1 and Hrt2 — that encode Hairy enhancer of Split (HES)-related transcriptional regulators (HRT) are major contributors of vessel formation [1220]. The combined loss of transcription factors HRT1 and HRT2 does not influence initial vasculogenesis, but it does affect subsequent development of major vessels.
10.6.2.2 Homeobox Transcription Factors
Homeobox (Hox) gene-encoded transcriptional factors are temporally and spatially restricted regulators of tissue patterning during embryogenesis owing to their DNA-binding homeodomain. The Hox regulators are involved in cell differentiation, proliferation, and migration. In particular they participate in endothelial cell fate, especially the transcriptional control of genes responsible for angiogenesis and vascular remodeling. The Hox transcription factors regulate genes involved in cell– and cell–matrix interactions, such as the expression of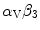 -integrin,matrix metallopeptidase MMP14, or urokinase plasminogen activator receptor [1221]. These proteins contribute to matrix remodeling during angiogenesis. The HOX genes are characterized by their clustered genomic arrangement. Each HOX gene can have up to 3 paralogs within independent clusters.
-integrin,matrix metallopeptidase MMP14, or urokinase plasminogen activator receptor [1221]. These proteins contribute to matrix remodeling during angiogenesis. The HOX genes are characterized by their clustered genomic arrangement. Each HOX gene can have up to 3 paralogs within independent clusters.
-integrin,matrix metallopeptidase MMP14, or urokinase plasminogen activator receptor [1221]. These proteins contribute to matrix remodeling during angiogenesis. The HOX genes are characterized by their clustered genomic arrangement. Each HOX gene can have up to 3 paralogs within independent clusters.Class-3 Hox factors (i.e., associated with 3 clusters, HoxA3, HoxB3, and HoxD3) have a pro-angiogenic role. Thay can provoke endothelial cell migration. Class-4 Hox factors (HoxA4–HoxD4) are involved in hematopoesis. Factor HoxB5 upregulates VEGFR2 receptor [1221]. In addition, in vitro, HoxB5 enhances endothelial cell sprouting and modulates the expression of adhesion molecules [1221]. Nevertheless, HoxB5 may mainly regulate intussusceptive vascular growth. In vivo, HoxB5 can upregulate angiopoietin-1 and -2 and VEGF [1221]. Its pro-angiogenic effect is abolished by soluble TIE2 (TIE2S), an angiopoietin antagonist. Factor HoxB5 also promotes the production ofmatrix metallopeptidase-1 and -2, but downregulates that of cell adhesion molecules such asβ3-integrin.
10.6.2.3 DLx and NKx Factors
Brain development relies on concomitant vasculature development. In mice, telencephalic angiogenesis is governed by a time and space (ventrodorsal) gradient determined by compartment-specific homeobox transcription factorsDistal-less homeobox protein DLx1 and DLx2,NK2 transcription factor-related homeobox protein NKx2-1, andpaired box protein Pax6 [1222]. These transcription factors also regulate the development of telencephalic neuroepithelial domains and neurons. Factor NKx2-1 is stimulated by sonic Hedgehog that influences both neurogenesis and angiogenesis. Distal-less homeobox DLx1 and DLx2 hamper the expression of Delta-like ligand-1, a Notch ligand. Paired box protein Pax6 hinders cornea vascularization.
10.6.2.4 ETV6 Transcriptional Repressor
Transcriptional repressor ETS-related translocation variant ETV634 is needed for sprouting of human endothelial cells. Factor ETV6 binds to the corepressor C-terminal-binding protein [1223]. The ETV6–CTBP complex temporally restricts VEGF-mediated pulse of Notch ligand DLL4. Control of DLL4 expression, which depends on the ETV6–CTBP complex, by VEGF is not iterated when VEGF signals continuously. Whereas VEGFR activation stimulates angiogenesis,Notch signaling primed by DLL4 inhibits the process. In endothelial cells, VEGF induces expression of DLL4 that then triggers the Notch pathway in adjacent cells to attenuate VEGFR activity and prevent spontaneous sprouting in the absence of sufficient VEGF signaling. The ETV6–CTBP complex further controls branching by regulating expression of other factors that limit angiogenesis, such asSprouty family members andVE-cadherin.
10.6.2.5 Nuclear Factor-κ B
Nuclear factor-κ B in endothelial cells has both positive and negative effects on angiogenesis [1224]. According to the context, NFκ B impedes or promotes apoptosis. Interactions of 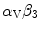 -integrin with matrix components that occur during migration of endothelial cells activate NFκ B factor.
-integrin with matrix components that occur during migration of endothelial cells activate NFκ B factor.
-integrin with matrix components that occur during migration of endothelial cells activate NFκ B factor.Nuclear factor-κ B upregulates VEGF expression as well as vascular endothelial growth inhibitor. Moreover, NFκ B can bind to angiostatic thrombospondin-1 and -2. In addition, NFκ B participates in activation of E-selectin in endothelial cells by CXCL4 chemokine.
Once NFκ B is activated byreactive oxygen species andtumor-necrosis factor-α, it induces expression of plasminogen activator inhibitor PAI1 and can hinder tissue-type plasminogen activator.35 On the other hand, NFκ B promotes expression of several matrixmetallopeptidases (MMP2, MMP3, and MMP9). Consequently, NFκ B can either favor or impede the degradation phase of angiogenesis. Last but not least, the activity of many angiostatic compounds depends on NFκ B activation.
10.6.2.6 PPARγ and PGC1α Coactivator
Vascular endothelial growth factor operates via bothPKCα and PKCβ to activatecAMP-responsive element-binding protein and subsequently produce the pro-angiogenic enzymecyclooxygenase-2 in endothelial cells. On the other hand,nuclear receptor NR1c3, or peroxisome proliferator-activated receptor PPARγ, precludes angiogenesis. Factor PPARγ suppresses membrane translocation of PKCα [1225].
Regular exercise improves blood circulation in the limbs, as it promotes the generation of new blood vessels in muscle. Numerous signaling pathways are activated during exercise that involve PP3 phosphatase, calmodulin-dependent kinases, AMP-activated protein kinase, stress-responsive P38MAPK, and reactive oxygen species. All of these pathways impinge on PGC1α [1226].
Transcriptional PPARγ coactivatorPGC1α elicits angiogenesis in muscles [1226].β-Adrenergic receptors increase expression of the Pgc1α gene and promote expression of vascular endothelial growth factor, thereby triggering angiogenesis. Factor PGC1α cooperates with the nuclear receptorestrogen-related receptor ERRα to regulate expression of the VEGF gene [1226].
In myocytes and adipocytes, the Pgc1α gene has an alternative promoter upstream from the proximal promoter that generates a PGC1α isoform with a few extra N-terminal amino acids. Changes in N-terminus of these proteins may confer specificity on the angiogenic program [1226]. Expression of mRNA from this promoter heightens 100-fold after exercise, whereas the transcription from the proximal promoter remains constant [1227].
10.6.2.7 Krüppel-like Factor
Krüppel-like factor KLF2 is produced in mature endothelial cells subjected to both shear stress and statins as well ascirculating pro-angiogenic cells, formerly calledendothelial progenitor cells, at a comparable level [1228]. It provokes endothelial functional differentiation. In mature endothelial cells, KLF2 prevents angiogenesis.
During senescence, KLF2 level decays [1228]. In aged mice, numbers of circulating SCA1 + , SCFR + , Lin − progenitors and SCA1 + , VEGFR2 + endothelial-committed progenitor cells lower. Moreover, KLF2 expression drops in spleen- and bone marrow-derived circulating pro-angiogenic cells.
10.6.2.8 Hypoxia-Inducible Factor
Hypoxia-inducible factor HIF1 intervenes in the compensatory angiogenesis to insufficient O2 supply. It initiates gene expression for numerous angiogenic regulators, vessel remodeling, as well as recruitment of bone marrow-derived,circulating angiogenic cells.
This ubiquitous, heterodimeric transcription factor mediates adaptive responses tohypoxia andischemia. Isotype HIF1 is made of O2-regulated HIF1α and constitutively expressed HIF1β subunits. Subtype HIF2α is an HIF1α paralog. Under hypoxia, HIF1α translocates to the nucleus, dimerizes with HIF1β, and binds to hypoxia response elements (HRE).
Transcriptional activity of HIF1 functions on genes that encode vascular endothelial (VEGF), placental (PlGF), and platelet-derived (PDGF) growth factors, stem cell factor, angiopoietin-2, and CXCL12 chemokine. These angiogenic factors bind to their cognate receptors (VEGFR1 and VEGFR2 for VEGF; VEGFR1 for PlGF; PDGFRα and PDGFRβ for PDGFB; SCFR for SCF; TIE2 for Ang2, and CXCR4 for CXCL12) in the plasma membrane of vascular endothelial cells, pericytes, and smooth myocytes.
In addition, HIF1 regulates the expression of hundreds of genes that encode, in particular, plasmalemmal receptors of angiogenic cytokines and homing signals for the recruitment of pro-angiogenic cells, such as endothelial progenitor cells, hematopoietic stem and progenitor cells, and mesenchymal stem cells, in addition to bone marrow-derived myeloid cells [1229].
10.6.2.9 HIF in Pulmonary Arterioles
Whereas systemic arterioles dilate in response to local tissue hypoxia to increase O2 delivery, pulmonary arterioles constrict in response to hypoxia to ensure proper local ventilation–perfusion ratios. Increased resistance of pulmonary arterioles results from elevated pulmonary arterial smooth myocyte tone as well as hypertrophy and proliferation. Agent HIF1 decreases the expression of voltage-dependentKV1.5 and KV2.1 channels and increases that oftransient receptor potential TRPC1 and TRPC6 channels andNa + –H + exchangers in pulmonary arterial smooth myocytes [1229]. Moreover, HIF1 triggers the production of endothelin-1 as well as angiotensin convertase and angiotensin receptor AT1 in human pulmonary artery fibroblasts.
10.6.2.10 Histone Deacetylases
Phosphorylated class-2Ahistone deacetylases recruit14-3-3 proteins for sequestration in the cytoplasm to overcome their repressor activity, because they cannot interact with their cognate transcription factors and corepressors. Dephosphorylation of class-2A HDACs allows nuclear import and gene activity repression.
Many kinases and phosphatase could phosphorylate and dephosphorylate HDAC7 according to location and signaling time for adequate and efficient cell reponses to stimuli, respectively.Phosphatase PP2 constitutively dephosphorylates HDAC7 histone deacetylase to control its function as a regulator of T-lymphocyte apoptosis and endothelial cell functioning during angiogenesis [1230].36
10.6.2.11 Histones
Histone-2A family member H2ax involved in DNA-damage repair promotes survival and proliferation of endothelial cells duringhypoxia and hypoxia-induced angiogenesis [1231]. Hypoxia leads to H2ax phosphorylation (H2axP or γ-H2ax) in endothelial cells, similarly to H2ax C-terminus phosphorylation that results from DNA damage by ataxia teleangiectasia mutated kinase (ATMK), ATM- and Rad3-related kinase (ATRK), and DNA-dependent protein kinase. Hypoxia-triggeredneovascularization requires functional H2ax in endothelial cells.
10.6.3 MicroRNAs
Post-transcriptional control of angiogenesis is ensured bymicroRNAs. MicroRNAs can be divided into 2 sets: pro- and anti-angiogenic microRNAs. Pro-angiogenic microRNAs include Let7, miR17-2, miR27b, miR126, miR130a, miR210, miR296, and miR378, whereas anti-angiogenic microRNAs encompass miR15b, miR16, miR92a, miR214, miR221, miR222, and miR328 [1232].
Dicer that produces final forms of microRNAs participates in postnatal angiogenesis induced by various stimuli. Growth factors can regulate microRNA expression. Factor VEGF regulates the expression of several microRNAs, such as components of the MyC cluster miR17-92 [1233].
Angiogenic sprouting of aortic arch vessels relies on the mechanosensitive transcription factorKrüppel-like factor KLF2a that provokes synthesis of endothelial-specific miR126 to activate VEGF signaling [1234]. MicroRNA-126 represses Sprouty-related EVH1 domain-containing protein SpRED1 and PI3Kr2 subunit, which prevent MAPK and PI3K signaling, respectively [886]. Therefore, miR126 derepressesPI3K andcRaf axes to support VEGF-induced angiogenesis (Table 10.13).
Table 10.13
MicroRNAs in angiogenesis (Source: [886, 1235]; SpRED: Sprouty-related EVH1 domain-containing protein).
Type | Targets |
|---|---|
MicroRNA-92a | Integrin-α5 (repression of angiogenesis) |
Sirtuin-1 (activation of Notch, repression of VEGF [?]) | |
MicroRNA-126 | PI3Kr2 (derepression of the PI3K axis) |
SpRED1 (derepression of the Raf1 axis) | |
IGFBP2 repression (inactivation of endothelial IGF1R) | |
Endothelial MerTK stimulation (cancer call MerTK inhibition) | |
MicroRNA-132 | RasA1 (derepression of Ras GTPase) |
MicroRNA-132 suppresses RasA1, a RasGAP, in the endothelium, thereby increasing Ras activity and promoting angiogenesis [1236]. Its expression is upregulated in a human embryonic stem cell model of vasculogenesis and in endothelia of human tumors and hemangiomas, but is undetectable in normal endothelium. On the other hand, RasA1 is produced in normal endothelium, but not tumor endothelium.
MicroRNAs encoded by the miR23–miR27–miR24 gene clusters37 are involved in cell cycle control, proliferation, and differentiation of various cell types. They abound in endothelial cells. Their pro-angiogenic effect results from the repression of the anti-angiogenic mediators Sprouty-2 and semaphorin-6A [1237].
Microvescicles represent a mode of communication between cancer and endothelial cells to launch endothelial cell migration. MicroRNAs are indeed packaged into and carried by microvescicles that can be taken up by endothelial cells. In cancer cells, some microRNAs such as miR126 can impede recruitment of endothelial cells [1235], whereas others such as miR9 are tumor promoters. In endothelial cells, upon delivery by microvesicles, miR9 decreases the abundance of SOCS5, an antagonist of the JAK-STAT pathway [1238]. Activated Janus kinase-1 and -2 phosphorylate signal transducer and activator of transcription STAT1 and STAT3.
10.6.4 Morphogens
Morphogens govern positions of various cell types in a developing tissue. These signaling substances build concentration gradients to generate and maintain the transcription of target genes at given concentration thresholds. Hence, genes may determine the anatomical structure of an organism [1239].38
10.6.4.1 Notch
Notch components mediate the arterial gene program,39 the venous specification via the nuclear receptor NR2f2,40 and the lymphatic commitment viaProspero homeobox gene product Prox1 [1167].41 Most lymphatics differentiate from veins.
The formation of endothelial tip cells at the leading edge of vascular sprouts is regulated by DLL4–Notch-1 signaling that is specific to the vasculature. Vascular-specific Delta-like ligand-4 regulates proliferation and differentiation of endothelial cells and vascular development. The Notch ligand DLL4 is expressed during vascular remodeling. Notch signaling initiated by DLL4 and restricted to the vascular system is involved in arteriovenous differentiation.
TheVEGF and Notch pathways are coupled. Signaling triggered by VEGFa provokes DLL4 production prominently in tip cells of endothelial sprouts. Notch ligand DLL4 induced by VEGF in tip cells activates Notch in adjacent endothelial cells, where it suppresses the expression of VEGFR receptors, thereby restraining excessive endothelial sprouting. In fact, expression of DLL4 in endothelial cells is controlled by VEGF that promotes Notch signaling to suppress the formation of additional tip cells [1240, 1241].
Notch signaling primed by DLL4 hence acts as a negative regulator of VEGF-induced angiogenesis. Blockade of interaction between DLL4 and Notch receptor prevents Notch signaling and inhibits angiogenesis intumors resistant to VEGF inhibitors [1242, 1243].42 The DLL4–Notch pathway intervenes during active vascularization rather than during vessel maintenance [1243]. Ligand DLL4 is upregulated intumor rather than in normal vessels [1242].
Sprouting blood vessels are organized with leader tip and follower stalk cells. Competition between endothelial cells for the tip position is regulated by glycosylation of Notch receptors and by the antagonism of Notch ligands Jag1 and DLL4 [1244]. In mice, whereas sprouting of endothelial tip cells is inhibited by the binding of Notch receptor to its ligand DLL4, the Notch ligand Jagged-1 is a potent angiogenic activator in cells that possess the Fringe family of glycosyltransferases. Notch glycosylation enhances DLL4-Notch signaling, whereas Jagged-1 has a weak signaling capacity.
Cerebral cavernous malformation protein CCM1 stabilizes endothelial junctions and keeps endothelium quiescent, thus acting as an anti-angiogenic substance [1245].43 It primes the inhibitory DLL4–Notch signaling to prevent endothelial cell proliferation, migration, apoptosis, and lumenogenesis. It then promotes PKB phosphorylation, but represses that of ERK kinases.
In arteries, in which Notch is more active, DLL4 was strongly expressed and VEGFR3 concentration is low. On the other hand, VEGFR2 lodges in both arteries and veins. In retinal tip cells, DLL4 expression is only weakly modulated by VEGFR2 signaling. Therefore, VEGFR2 is not essential for DLL4 expression in tip cells [1246]. Moreover, activated Notch is a potent inhibitor of VEGFR3 synthesis, but not of VEGFR2 expression. Receptor VEGFR2 is indeed not or only weakly regulated by Notch [1246]. Activity of VEGFR3 is pro-angiogenic in endothelial cells, in which Notch signaling is weak or absent. In low Notch activity, VEGFR3 upregulation allows ligand-independent, excessive, deregulated angiogenesis even in the absence of VEGF–VEGFR2 signaling.
Notch and VEGFR2 are antagonists in the regulation of VEGFR3 level. Notch regulates VEGFR3 activity independently of VEGFR2 receptor. Conversely, VEGFR3 kinase activity enables Notch-regulated sprouting. On the other hand, VEGFR2 strongly improves VEGFR3 density (but not DLL4) at the angiogenic front. Therefore, VEGFR3 may have an active, ligand-dependent, pro-angiogenic and a Notch-mediated, passive signaling modes [1246].
The non-canonical Notch ligand Delta-like homolog DLk1,44 a transmembrane glycoprotein expressed in the endothelium that lacks the DSL domain, impedes angiogenic sprouting, endothelial cell migration, and tubulogenesis via Notch receptors [1247].
Transmembrane syndecans are heparan sulfate proteoglycans that function as coreceptors for growth factors. They are implicated in angiogenesis. Syndecans are implicated in communication between endothelial and smooth muscle cells aimed at ensuring proper structure and function of blood vessels and their maintenance. These mural cell types communicate via, at least partly, Notch-3 and syndecan-2 plasmalemmal receptors. The latter is produced in smooth myocytes upon contact with endothelial cells and Notch activation in smooth myocytes [1248]. Both Notch-2 and -3 contribute to the upregulation of syndecan-2. In particular, syndecan-2 interacts with Notch-3 and regulates expression of several Notch-3 target genes (only in coculture conditions). Conversely, expression of syndecan-2 in smooth myocytes needs Notch-2 and Notch-3 proteins.
10.6.4.2 Wnt
The Wnt–β Ctn pathway favors DLL4 expression in the vasculature. Conversely, the DLL4–Notch complex activates Wnt in stalk cells. In addition, Notch promotes vascular stability via Notch-regulated ankyrin repeat-containing protein (NRARP) [886]. Agent NRARP limits Notch activity, but stimulates Wnt signaling to stabilize stalk cells.
The planar cell polarity mode of Wnt signaling participates in endothelial cell proliferation and angiogenesis. On the other hand, Disheveled-associated activator of morphogenesis DAAM1 that operates as a formin to cause actin polymerization and especially microtubule stabilization precludes endothelial cell proliferation and migration [1249].
Retinal myeloid cells (macrophages) produce Wnt ligands to suppress retinal (postnatal) angiogenesis via the non-canonical Wnt–VEGFR1 pathway [1250].
10.6.4.3 Hedgehog
Hedgehog signaling participates in blood vessel development. It regulates distinct vascular patterning events using VEGF-dependent and -independent mechanisms [1251]. Expression ofVEGF in the mouse embryo depends on Hh signaling. Hedgehog activity in both endothelial tube formation and Notch-dependent arterial identity solely depends on VEGF regulation. Hedgehog effects are limited by Hh inhibitor Patched-1 that depends neither on VEGF nor Notch.
10.6.5 Growth Factors
Many angiogenic factors exert chemotactic and mitogenic activities on vascularendothelial andsmooth muscle cells as well as on fibroblasts. Moreover, they control the metabolic functions of these cells. Numerous growth factors act on endothelial and/or smooth muscle cells (Table 10.14).
Table 10.14
Examples of growth factors and cytokines involved in angiogenesis and arteriogenesis (Source: [1252–1254]). Thymidine phosphorylase (TymP; a.k.a. gliostatin and platelet-derived endothelial cell growth factor [PDECGF or ECGF1]) that belongs to the family of glycosyltransferases stimulates endothelial cell growth and promotes angiogenesis.
Molecule | Target |
|---|---|
Angiogenin | EC |
Angiopoietin-1 | EC |
CCL2 | EC, SMC |
EGF | EC |
FGF | EC, SMC |
Interleukin-1 | EC, SMC |
Interleukin-20 | EC |
PDGF | EC, SMC |
TGF | EC (TGF  , TGF , TGF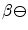 ), SMC, ECM ), SMC, ECM |
TNF | EC  |
TymP | EC, SMC |
VEGF | EC |
Certain growth factors, such as vascular endothelial growth factor and angiopoietin-1, can act both on angiogenesis and vascular permeability. Transforming growth factor-β stimulates extracellular matrix production to stabilize the structure of new vessels.
10.6.5.1 Vascular Endothelial Growth Factors
Vascular endothelial growth factors regulate the initial stages of blood vessel formation. Later, they are also implicated in association with other activators, such as angiopoietins, fibroblast (FGF), insulin-like (IGF), and transforming (TGFβ) growth factors, as well as TNFα, apelin, ephrin, and Notch. Most of these factors act on other cell types and complement or coordinate VEGF signaling rather than operating as independent regulators of angiogenesis.
In the absence of TGFβ1, VEGF favors endothelial cell survival via VEGFR2 and P38MAPK [1255]. In the presence of TGFβ1, VEGF promotes cell apoptosis. During angiogenesis, apoptosis is required for pruning the forming vascular network. Factor TGFβ1 promotes the expression of FGF2 by endothelial cell, which upregulates the expression of VEGF and acts viaERK1 and ERK2 kinases.
Vascular endothelial growth factor is a potent mitogen for endothelial cells derived from arteries, veins, and lymphatics [1256]. Moreover, VEGF stimulates cardiomyocytes. Factor VEGF exerts its angiogenic effects via its cognate VEGFR receptors (Fig. 10.1). Endothelial cells activated by VEGFa, assemble, and form tubes.