(1)
Project-team INRIA-UPMC-CNRS REO Laboratoire Jacques-Louis Lions, CNRS UMR 7598, Université Pierre et Marie Curie, Place Jussieu 4, 75252 Paris Cedex 05, France
Abstract
Vascular endothelium constitutes the interface between the flowing blood and the deformable solid wall. The endothelium is a thin layer of connected and anchorage-dependent cells that are subjected to chemical, physical, and mechanical stimuli. They are directly exposed to molecules that circulate in the blood stream.
Vascular endothelium constitutes the interface between the flowing blood and the deformable solid wall. The endothelium is a thin layer of connected and anchorage-dependent cells that are subjected to chemical, physical, and mechanical stimuli. They are directly exposed to molecules that circulate in the blood stream.
Vascular endothelium has several functions, as it is involved in: (1) blood–wall exchange control; (2) vasomotor tone modulation;1 (3) coagulation regulation; (4) vessel wall growth and remodeling; and (5) inflammation and immune defense owing to leukocyte adhesion and transmigration [841]. Last, but not least, vascular endothelium is required for angiogenesis (Sect. 10.2). During angiogenesis, the arteriovenous differentiation drives vessel maturation.
Although endothelial cells of blood and lymph vessels share many features (strong apicobasal polarity and expression of certain endothelial markers), they are specialized according to vessel function and convected fluid loading. The endothelium of terminal lymphatics lacks a continuous basement membrane and intercellular spaces are not tightly sealed by junctional complexes.
Fluorescent plant virus can be used to image small-bore vessels of the macrocirculation in deep tissues using fluorescence microscopy [842], as virus is incorporated in vascular endothelial cells. This technique identifies arterial and venous compartments, as fluorescent virus uptake in the veins occurs at a much higher rate than in the arteries.
The semi-permeable endothelial barrier acts in molecule exchange between blood and vessel wall or interstitial space of perfused tissues (Sect. 9.6), thereby regulating tissue fluid homeostasis. Endothelial permeability of transported molecules depends on molecular size as well as intercellular junction nature and pattern. This size-selective transport governs fluid balance of tissues.
Vascular endothelium determines the vasomotor tone as well as growth and proliferation of vascular smooth myocytes via the release of several compounds (Sect. 9.10). Vasoactive substances include vasoconstrictors such as endothelin-1 and vasodilators such as nitric oxide. In particular, NO gas is produced by nitric oxide synthases, especially endothelial NOS3 isoform. Messenger NO diffuses from vascular endothelial cells to smooth myocytes, where it activates NO-sensitive soluble guanylate cyclase to initiate signaling.
Vascular endothelium regulates blood coagulation as well as thrombolysis (Sect. 9.8). Furthermore, it controls adhesion and extravasation of flowing leukocytes (Sect. 9.7), thus inflammation and immune defense. In normal conditions, the vascular endothelium has anti-inflammatory and -thrombotic activities. It responds by synthesizing multiple molecule types. In particular, endothelial adenosine triphosphate diphosphohydrolase hydrolyzes ADP and ATP into AMP molecule.
Endothelial cells ensure formation of the blood vessel network into a hierarchical set of arteries, arterioles, capillaries, venules, and veins that enables the transport of fluid, nutrients, circulating cells, hormones, and gasses to organs.
Endothelial cells detect hemodynamic stresses via mechanosensors, such as adhesion molecules (mainly integrins), ion channels, and plasmalemmal receptors (GPCRs and RTKs). Signaling pathways (MAPK, PKB, PKC, and ROS) augment the activity of transcription factors (Activator proteins AP1 and AP2,cAMP response element,early growth response protein EGR1, andNFκB) with a magnitude that depends on the cell type, i.e., vascular region.
Time-dependent hemodynamic stresses applied on and within the vessel wall (wall shear stress, axial and circumferential tensions within the wall) are implicated in: (1) the secretion of vasoactive substances (nitric oxide, endothelin, and prostacyclin, among others) and regulation of the vascular tone that determines the vessel bore according to the magnitude of sensed mechanical stresses; (2) short-term wall adaptation and long-term remodeling, as they influence cell signaling that directs cell growth (growth inhibitor heparin and growth factors such as platelet-derived growth factor), differentiation, migration, and apoptosis; (3) expression of proteins entailed in coagulation and fibrinolysis (tissue plasminogen activator, plasminogen-activator inhibitor, tissue factor, etc.), in cellular adhesion (vascular cell adhesion molecule VCAM1 and intercellular adhesion protein ICAM1), in diapedesis (CCL2 chemokine); and (4) vasculature diseases because they affect the cell functioning and transport processes.
Wall shear stress, in particular, upregulates the connexin expression and entail calcium influx that activate protein kinases and nitric oxide synthase for fast nitric oxide release.
9.1 Endothelial Cell
Endothelial cells are flat with a central, elongated, projective nucleus that yields a wavy wetted surface at the microscopic level. The endothelium surface, either from fresh arterial walls mounted on an appropriate holder and kept in physiological buffer or from cultures, can be assessed by a scanning force microscope associated with a phase-contrast microscope [843].
In addition to endothelial cells that cover blood and lymph vessels, mature endothelial cells (circulating endothelial cells [CEC]) as well as endothelial progenitor cells (circulating endothelial progenitor cells [CEPC]; Sect. 9.3) circulate in the blood flow at a very low concentration. Resident endothelial cells may detach during the normal turnover after apoptosis, with clearance by the reticuloendothelial system, in the absence of endothelial damage. Circulating endothelial progenitor cells are able to form patches at sites of endothelial discontinuity to ensure the integrity of the vessel wall [844].2
Flattened endothelial cells (thickness 1–2 μm, width 10–15 μm, length in the streamwise direction 60–100 μm) have rest and stretched (flow-adapted) configurations. The height variation due to projective nucleus has been measured along the endothelium with a maximum of 750 nm.
Observed plasmalemmal granules and ring-like structures of various sizes have been assumed to be associated to the cytoplasmic layer of the plasma membrane rather than the outer one. Moreover, fibers can be seen, likely associated with actin filaments, that are bound to the cytoplasmic face of the plasma membrane.
The cell membrane is covered by a thinglycocalyx. The between-cell space width ranges from 10 to 20 nm withtight andgap junctions.
Endothelial cells contain Weibel-Palade bodies, long rod-shaped storage organelles (length 1–5 μm, caliber ∼ 200 nm). These storage and secretory granules are filled with von Willebrand factor.3 The von Willebrand factor recruits platelets to the site of injury.4 The architecture of the Weibel-Palade body and tubular folding of von Willebrand factor requires a low pH [846]. The tubules must not be disassembled prior to exocytosis. Once released, von Willebrand factor unfolds rapidly and efficiently at neutral pH to trap circulating platelets, as it forms platelet-catching filaments (length ∼ 100 μm). Weibel-Palade bodies also contain P-selectins.
Endothelial cells monitor cell internal state as well as environmental actions. Endothelial cells especially sense hemodynamic and hormonal stimuli and respond by secreting various mediators. Endothelial cells experience blood pressure, axial and circumferential tension from connecting endothelial cells, and blood friction on the wetted surface, i.e., the wall shear stress5 (WSS). The applied forces are unsteady with noticeable spatial and temporal magnitude gradients as well as possible direction changes. The stress distributions in the membrane and cytosol affect endothelial functions.
9.1.1 Glycocalyx
The glycocalyx forms a thin layer6 between the circulating blood and endothelium. It is a hydrated mesh of negatively charged glycosaminoglycans,7 proteoglycans, glycoproteins, and glycolipids secreted by endothelial cells.
The glycocalyx is the first barrier to molecular transport from the flowing blood to the vessel wall, providing hydraulic resistance to mass transport through this sieve [847]. The transport conductance in the glycocalyx depends on molecular size.8 Both the hydrostatic and osmotic pressures act on transport across the glycocalyx. However, albumin (molecular mass 67 kDa) and fibrinogen (molecular mass 340 kDa) cross the glycocalyx at about the same rate. Charge restriction imposed by the glycocalyx also determine accessibility of proteins. Nonetheless, the glycocalyx generates nanodomains associated with a heterogeneous distribution of negative charges that modulate transendothelial transport.
At the basal state, the rheological properties of the glycocalyx might induce a lift that prevents cell adhesion [848]. In inflammatory sites, heparan sulfate on the surface of endothelial cells is a potential ligand for P-selectins (expressed by endothelial cells) and L-selectins (expressed by leukocytes), which are involved in initial attachment and rolling of leukocytes on the endothelium [849]. It also binds chemokines for stable adhesion of leukocytes on the endothelium.
The glycocalyx, which is a polyelectrolyte coating, has been modeled as a semi-infinite, doubly periodic array of parallel charged cylinders [850]. Only the luminal layer part of the glycocalyx model markedly influences transport.
Nacetylglucosamine is one of the main components of glycocalyx oligosaccharides. It interacts with endothelial Glc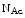 -recognizing lectins of the luminal surface, among which some may participate in flow sensing such asglycosylated endothelial Na + channel (ENaC) [851]. Connection of hyaluronan to lectin-bearing substance such as ENaC channel.
-recognizing lectins of the luminal surface, among which some may participate in flow sensing such asglycosylated endothelial Na + channel (ENaC) [851]. Connection of hyaluronan to lectin-bearing substance such as ENaC channel.
-recognizing lectins of the luminal surface, among which some may participate in flow sensing such asglycosylated endothelial Na + channel (ENaC) [851]. Connection of hyaluronan to lectin-bearing substance such as ENaC channel.9.1.2 Endothelial Cell Adhesions
Endothelial intercellular junctions are necessary for formation and integrity of the interface between blood and vessel wall. The main junctions between endothelial cells are adherens and tight junctions that are composed of transmembrane adhesion molecules linked to cytoskeletal-binding proteins and intracellular signaling partners.
Intercellular junctions not only yield attachment sites, but also transfer signals for morphogenesis and stabilization of the vessel wall architecture. Vascular homeostasis relies also on cellular adhesion with the extracellular matrix.Integrins link endothelial cells with constituents of the extracellular matrix, such as fibronectin or vitronectin. Their cytosolic domains are associated with actin cytoskeleton via talin and vinculin.
9.1.2.1 Junctions between Endothelial Cells
Intercellular junctions include tight, adherens, and gap junctions. Adherens junctions are mainly required for correct vasculo- and angiogenesis and remodeling, whereas tight junctions essentially control the endothelial barrier. Endothelial cells do not have desmosomes. In addition,α2β1– and α5β1-integrins have also been identified in endothelial clefts.
In endothelial cells, actomyosin filament contraction can generate forces (up to 120 nN) that pull perpendicularly to the face of intercellular contact, the so-calledtugging forces.Mechanical loading at cell–matrix and –cell adhesions causes focal adhesion growth. In particular, the size of adherens junctions enlarges when tugging forces rises and conversely [852]. Actomyosin-dependent regulation of adherens junction size is supported by smallRac1 GTPase.
Adherens Junctions
Adherens junctions are mainly composed ofcadherins (Vol. 1 – Chap. 7. Plasma Membrane). Cadherin cytoplasmic tail contains 2 binding sites. The first binding site associates withβ- and γ-catenins in a mutually exclusive manner. The second links to catenin-δ1, an inhibitor of Rho GTPases. Cadherin then indirectly connects to α-catenin via β- or γ-catenin. Catenin-α links the cadherin–catenin complex to the actin cytoskeleton. Vascular endothelial (or Cdh5) and possibly neuronal (or Cdh2) cadherins thus bind to intracellular partners that contribute to signaling and dynamics of the actin cytoskeleton.9 Catenin-γ10 can also tether cadherin-5 tointermediate filaments viadesmoplakin to form the complexus adhaerentes, a special type of cell junctions. The desmoplakin–vimentin complex corresponds to an additional agent of mechanical stability.
Catenins-β are able to bind to junctional proteins, such asIQ motif-containing GTPase-activating protein IQGAP1, platelet–endothelial cell adhesion molecule PECAM1, casein kinase-2, as well as signaling and transcription factors, such asWnt,adenomatous polyposis coli Ub ligase, andT-cell factor [854]. Scaffold IQGAP1 not only links to β-catenin and E-cadherin, but also to small GTPasesCDC42 andRac, as well as actin,calmodulin, and microtubule-associated cytoplasmic linker integral protein CLIP170.11Adhesion molecule PECAM1 that is concentrated in endothelial clefts interacts with Tyr-phosphorylated β-catenin andphosphatase PTPn11. Molecule PECAM1 may participate in modulating adherens junction assembly and restoring endothelial barrier integrity after injury. In addition, PECAM1 bindsαVβ3-integrins and regulates the function of α4β1– and β2-integrins to possibly mediate transendothelial migration of leukocytes.
α-Catenin that links to β-catenin and actin-polymerizing proteins, such as α-actinin, vinculin, vasodilator-stimulated phosphoprotein, and formin, as well as actin microfilaments, promotes actin bundling, thereby stabilizing adherens junction and cleft. Furthermore, actin polymerization is needed for adherens junction assembly. Non-muscle myosin heavy chain-2A is an another regulator of adherens junction formation [854]. Cadherin-5 can regulate intercellular permeability by modulating GTP binding to and GTP hydrolysis of small GTPases CDC42, Rac, andRhoA [854]. Cadherins can interact with actin-related proteic ARP2–ARP3 complex that associates with Wiskott-Aldrich syndrome protein (WASP), cortactin, and vinculin. Wiskott-Aldrich syndrome protein is an effector of CDC42 GTPase. The CDC42–ARP2/3–WASP pathway increases actin polymerization. Cadherin-5 can also activate GTPase Rac via Rac-specific GEF T-cell lymphoma invasion and metastasis Tiam1. By activating small GTPases, cadherins can control actin polymerization at intercellular junctions and modulate paracellular permeability.
Catenin-δ1 regulates cadherin-5 expression and insertion into the plasma membrane. The Ctnnδ1–Cdh5 complex precludes binding of ubiquitin ligase Hakai, hence preventing cadherin-5 degradation. Catenin-δ1 also has many partners, such as microtubule nanomotor kinesin and regulatory kinases and phosphatases (e.g., SRC kinase family member Fyn, FRK family kinase Fer, and plasmalemmalPTPRm and cytosolicPTPn6 protein Tyr phosphatases) [854]. Unlike β-catenin and plakoglobin, catenin-δ1 does not associate with the actin cytoskeleton, but with microtubules. However, catenin-δ1 regulates the contractile apparatus of endothelial cells and modulates endothelium permeability. The Ctnnδ1–Cdh5 complex actually impedes the activity of RhoA GTPase that mediatesmyosin light chain phosphorylation and actin stress fiber formation.
Several receptor and cytoplasmic protein Tyr phosphatases localize to adherens junctions and can dephosphorylate components of the cadherin–catenin complex and controlRho activity.
Vascular endothelial protein Tyr phosphatase PTPRb restricted to the endothelium associates specifically with and dephosphorylates VE-cadherin. Moreover, the PTPRb–Cdh5 complex can contribute to strengthening of adherens junction barrier by a phosphatase-independent mechanism [854].
Phosphatase PTPRj12 abounds in endothelial cells at least in arteries and capillaries of several organs [853]. When it associates with the Cdh5–βCtn complex, it dephosphorylatesVEGFR2 vascular endothelial growth factor receptor.
Receptor-like protein Tyr phosphatase PTPRm is strongly present in endothelial junctions of arteries and continuous capillaries. Phosphatase PTPRm dephosphorylates catenin-δ1, thus modulating interaction of catenin-δ1 with cadherin-5 and controlling catenin-δ1 regulation of RhoA activity.
Protein Tyr phosphatase PTPn11 is an additional component of adherens junctions that links to VE-cadherin. Dissociation of PTPn11 can expose junctional proteins to phosphorylation by kinases and cause adherens junction disassembly. In addition, PTPn11 prevents small GTPase RhoA activity to stabilize adherens junction.
Small GTPasesCDC42,Rac, and RhoA contribute to the regulation of adherens junctions. Bradykinin, histamine, platelet-activating factor, and thrombin that heighten endothelial permeability provoke disassembly of adherens junctions via RhoA GTPase. On the other hand, Rac GTPase promotes endothelial barrier by stabilizing adherens junctions. Moreover, activated CDC42 is involved in reformation of adherens junctions in endothelial cells during recovery from abnormal permeability induced by permeability-increasing agents. Small GTPases CDC42 and Rac modulate interactions between α-catenin and the cadherin–catenin complex to favor formation of adherens junctions. Activated CDC42 and Rac indeed interact with β-catenin-sequestering protein IQGAP to free β-catenin that can then binds with its partners cadherin and α-catenin [854].
Permeability-increasing factors, such as thrombin andVEGF, induce phosphorylation of cadherin, β-catenin, and catenin-δ1 to disrupt adherens junctions [854]. Phosphorylation bySrc orPKC of adherens junction constituents can modify affinity of catenins for cadherin-5, as well as Cdh5–actin interactions. Conversely, phosphatases stabilize adherens junctions. PTPn11 Phosphatase associates with and protect the cadherin–catenin complex.
Tight Junctions
Tight junctions containoccludin,junctional adhesion molecules, andclaudins that interact directly or indirectly with cytoplasmic partners, such ascingulin andzonula occludens adaptors. Zonula occludens proteins (ZO1–ZO3) are members of the family of membrane-associated guanylate kinases (MAGUK). Zonula occludens proteins and cingulin contribute to interaction of tight junction with the actin cytoskeleton.
Occludins form homotypic bonds. Their cytoplasmic C-terminus associates with zonula occludens protein ZO1, hence with the actin cytoskeleton to stabilize tight junction. Arterial and blood–brain endothelial barriers that are the least permeable of the vasculature contain a much greater number of occludins than do other compartments.
Among 24 known members of the claudin family, only claudin-5 is specifically expressed by endothelial cells. Yet, several claudins are synthesized in endothelial cells (claudin-1, -3, -5, and -12). Claudin-5 that is particularly produced by cerebral endothelial cells is a major regulator of the blood–brain barrier function. Claudin extracellular regions form homo- and heterotypic bonds. Its cytoplasmic part also binds to zonulaoccludens protein ZO1, and therefore indirectly to ZO1 partners.
Junctional adhesion molecules, endothelial cell-selective adhesion transmembrane glycoproteins, and coxsackievirus and adenovirus receptors associate with tight junctions, but do not build strands per se [853]. However, they modulate leukocyte diapedesis across the endothelium.
Junctional adhesion molecules comprise 3 main types: JAM1 (or JAMa), JAM2 (or JAMb), and JAM3 (a.k.a. JAMc and veJAM). Isotype JAM1 resides in epithelial and endothelial cells; JAM2 in high endothelial venular cells, i.e., endothelial cells of postcapillary venules of lymphoid tissues that form the leakiest endothelium of lymphatic vessels; JAM3 exclusively in endothelial cells. Junctional adhesion molecules are able to bind partitioning-defective protein Par6, small GTPaseCDC42, andPKCζ, thereby recruiting these signaling mediators to tight junctions.
Zonula occludens proteins interact directly or indirectly via bridging proteins with claudins, occludins, and junctional adhesion molecules. They intervene in spatial organization of tight junction constituents, particularly occludins. Zonula occludens proteins link tight junction proteins to the actin cytoskeleton and recruit signaling molecules. In addition, ZO1 also binds to adherens junction protein α-catenin, gap-junction component connexin-43, and actin-polymerizing proteins vasodilator-stimulated phosphoprotein and spectrin, hence linking tight junctions to the actin cytoskeleton. Therefore, in endothelial cells, adherens and tight junctions that are intermingled interact via common partners such as ZO1 for their formation, maintenance, and remodeling. In addition,nectins and their intracellular partners such asafadin contribute to the organization of adherens and tight junctions.
Endothelial barrier requires endothelial markers cadherin-5 and claudin-5 of adherens and tight junctions, respectively. Homotypic Cdh5-based adhesions control claudin-5 expression, as they prevent the nuclear accumulation of transcriptional repressorsFoxO1 and β-catenin that inhibit claudin-5 promoter [855].13 Thiscrosstalk relies on Z01 and JAMs. The crosstalk is mutual as tight junction molecules such as junctional adhesion molecules can regulate cadherins in endothelial cells.
The RhoA–RoCK pathway that causes actin stress fiber formation can then induce a loss in junctional occludin, disrupt tight junctions, and increase endothelial permeability. Histamine, lysophosphatidic acid, thrombin, and vascular endothelial growth factor disassemble tight junctions via the RhoA–RoCK pathway that phosphorylates occludin and ZO1, possibly via kinaseSrc,protein kinase-C, andcasein kinase-2. Conventional PKC isoforms (α, β, and δ) also phosphorylate junctional adhesion molecules.
Gap Junctions
Each gap junction is made up of 2 connexons that correspond to contribution of each of the 2 partner cells. Connexon is constituted of 6 connexins. The intercellular pore formed by connexons (caliber ∼ 2 nm) has an open or closed configuration. Gap junction gating is regulated by connexin phosphorylation. Both Ser–Thr and Tyr phosphorylation of connexins induce channel closure. Phosphorylation also regulates the rate of gap junction assembly and turnover.
Endothelial cells express connexins Cx37, Cx40, and Cx43. As connexins form gap junctions, they allow rapid exchange of low-molecular-mass messengers, such as calcium ions and inositol trisphosphate, between contiguous cells. Junction protein ZO1 binds connexin-43 and facilitates communication between tight, adherens, and gap junctions.
Connexins contribute to nitric oxide production and endothelial barrier integrity. Moreover, endothelial Cx43 can also associate with Cx40 of neutrophils during their extravasation and vascular smooth myocytes.
9.1.2.2 Cell–Matrix Junctions
Focal adhesions are sites of adhesions of endothelial cells to the extracellular matrix mediated byintegrins. Endothelial cells express mainly on their abluminal surface numerous heterodimeric integrins (α1β1, α2β1, α3β1, α5β1, α6β1, αVβ3, α1β5, and αVβ5) [854]. These transmembrane glycoproteins interact with matrix proteins, such as fibronectin, fibrinogen, vitronectin, and collagen. Association between integrins and extracellular matrix constituents restricts the passage of macromolecules across the endothelial barrier.
Integrin cytoplasmic domain connects to actin-binding proteins, such as α-actinin, filamin, paxillin, talin, tensin, vinculin, and zyxin.α-Actinin that binds zyxin targets vasoactive stimulatory phosphoprotein and profilin.Filamin links to small GTPasesCDC42,Rac,RhoA, andRal1, as well as RhoA-associated kinase,Trio with its 3 enzymatic domains,14 and caveolin-1.Paxillin interacts with P21-activated kinase (PAK) andPAK-interacting exchange factors RhoGEF6 and RhoGEF7,Abelson Tyr kinase (Abl), andRas GTPase-activating protein RasA1.Tensin binds multiple phosphotyrosine proteins, such asBCAR1 (or CAS) andSrc kinase.Vinculin interacts with theactin-related proteic ARP2–ARP3 complex andphosphatidylinositol 4-phosphate 5-kinase. These proteins coordinate signals between focal adhesions and the actin cytoskeleton.
These proteic interactions define theadhesome, a signaling platform integrator of many signaling axes. Adhesome regulates actin polymerization and focal adhesion function. For example, ligation of αVβ3-integrin with matrix proteins causes: (1) tyrosine phosphorylation of focal adhesion kinase, paxillin, cortactin, and ezrin; (2) Ca2 + influx; and (3) activation ofphospholipase-A2 andRac GTPase.
Focal adhesion constituents are recruited into focal adhesions upon tyrosine phosphorylation byfocal adhesion kinase andSrc kinases. Integrin clustering provokes FAK autophosphorylation (activation). Kinase Src further phosphorylates activated FAK enzyme. Activated FAK phosphorylates various substrates, such as paxillin, tensin, PI3K, and BCAR1, that aggregate to form focal adhesions.
Paxillin recruits other focal adhesion constituents, as it associates with vinculin,CRK,C-terminal Src kinase, and Src, as well as ArfGAPs APAP1 and APAP2 [854]. Activated APAP1 regulates CDC42 and Rac, as it can complex with RhoGEF6 and RhoGEF7 that serves as CDC42- and Rac1-GEF to maintain or restore endothelial barrier.
Focal adhesion kinase not only associates with paxillin and talin, but also with SRC family kinases Src and Fyn, guanine nucleotide-exchange factor RasGRF1, growth factor receptor-bound protein GRB2, adpribosylation factor GAP-containing, SH3, ankyrin repeats, and PH domain protein ASAP1, BCAR1 adaptor, RhoGAP26, and actin-polymerizing proteins ezrin and WASP [854]. Focal adhesion kinase is phosphorylated (activated) in response to integrin activation.
Various mediators, such asthrombin,histamine, hydrogen peroxide, andVEGF, are able to cause FAK phosphorylation and promote focal adhesions to maintain endothelial barrier integrity. Furthermore, focal adhesion kinase restores endothelial barrier function after intervention of inflammatory mediators that causes cell junction disassembly.
On the other hand, increased FAK activity allows hyperosmolarity-induced strengthening of adherens junctions. Focal adhesion kinase hinders activity of GTPase RhoA, as it associates with RhoA inhibitors RhoGAP26 and P190RhoGAP. Furthermore, focal adhesion kinase activates APAP1 protein. In addition, FAK is required for normal vascular development.
Kinases of the SRC family can be stimulated by activatedintegrins. Activated Src kinase interacts with focal adhesion kinase for full kinase activity. Among SRC family members, Src kinase contributes to increased endothelial permeability in response to superoxide anion (O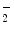 ), thrombin, andVEGF [854].15 Kinase Src not only elevates endothelial junction permeability, but also caveola-mediated transcytosis.
), thrombin, andVEGF [854].15 Kinase Src not only elevates endothelial junction permeability, but also caveola-mediated transcytosis.
), thrombin, andVEGF [854].15 Kinase Src not only elevates endothelial junction permeability, but also caveola-mediated transcytosis.Proline-rich Tyr kinase PYK2, a Ca2 + -dependent cytosolic kinase, also bindsintegrins. It is highly expressed in pulmonary endothelial cells. It is rapidly phosphorylated upon angiotensin and mechanical stimuli. It could regulate endothelial barrier by activating RhoA GTPase. Activated RhoAGTP participates in the formation of focal adhesions via its effectors RoCK andDiaphanous as well as PIP5K recruitment.
Transmembrane domains of integrins also interact withtetraspanins, GPI-anchored urokinase-type plasminogen activator receptor, and caveolin-1.Caveolin-1 is involved in formation of focal adhesions, development of basement membrane, paracellular endothelial permeability, and integrin signaling [854].
9.1.2.3 Myoendothelial Junctions
Myoendothelial junctions (MEJ) correspond to endothelial projections that protrude and cross holes of the endothelialbasement membrane and internal elastic lamina to reach adjacent smooth myocyte membranes.
The principal regulators of vascular tone are neural, endothelial, and mechanical stimuli that initiate vasodilation or vasoconstriction. Three primary vasodilatory signals include messengers produced by endothelial cells, nitric oxide and prostaglandins, and endothelium-dependent vasodilatory hyperpolarization. The latter refers to the transfer of an endothelium-derived electrochemical current through direct coupling between endothelial and smooth muscle cells via myoendothelial gap junctions (contact mechanism) and activity of ion carriers located in myoendothelial microdomains (diffusible endothelial factor release; Table 9.1).
Table 9.1
Mechanisms of endothelium-derived hyperpolarization (Source: [856]). Endothelium-derived hyperpolarization (EDH) relies, at least partly, on cytosolic calcium influx upon liganded receptors and/or hemodynamic stresses and activation of potassium channels, such as small, intermediate, and, in some cases, large-conductance calcium-activated potassium channels (EC: endothelial cell; MEES: myoendothelial microdomain extracellular space; SMC: smooth myocyte.
Diffusible factors | |
|---|---|
Potassium ion | Efflux from EC to MEES through KCa |
(transient, localized cue) | and activation of SMC Na + –K + ATPase |
and KIR channel | |
Epoxyeicosatrienoic acids | Opening of EC and SMC KCa1.1 (BK), |
(EETs) | Ca2 + entry through TRPV4 |
Hydrogen peroxide | Vasoconstriction, activation of |
(H2O2) | SMC KCa, KATP, |
and Na + –K + ATPase; no effect on EDH | |
in human radial and subcutaneous arteries | |
C-type natriuretic peptide | Vasorelaxation, activation of NP2 |
(CNP) | and NP3 receptors; role questionable |
Contact-mediated mechanisms – Gap junctions | |
Electrochemical coupling | Eventual modulation by K + , EETs, |
H2O2, CNP | |
Myoendothelial projections (MEP) contain myoendothelialgap junctions composed ofconnexins, thereby enabling direct signaling between vascular endothelial cells and smooth myocytes. Endothelial hyperpolarization is directly transmitted to adjacent smooth myocyte via the myoendothelial gap junction. Endothelium-dependent hyperpolarization of adjacent smooth myocytes closes theirCaV1.2 channel.
Hydrogen peroxide (H2O2) can influence gap junctional coupling in addition to modulating the sensitivity of the contractile apparatus to calcium and activating smooth muscle Na + –K + ATPase and BKCa and KATP channels [856]. However, H2O2 does not play a significant role in endothelium-dependent hyperpolarization [857].
Endothelium-dependent hyperpolarization factors (EDHF) comprise actions of K + ion, nitric oxide, prostaglandins, cytochrome-P450 products epoxyeicosatrienoic acids, and myoendothelial electrical coupling, but is neither prostacyclin nor nitric oxide.
Two types of Ca2 + -activated K + channels are involved in endothelium-dependent hyperpolarization (Table 9.2): (1) small conductance Ca2 + -activated K + channels (SK or KCa2.3) that are widely distributed over the endothelial plasma membrane and (2) intermediate conductance Ca2 + -activated K + channels (IK or KCa3.1) that mainly localize to the myoendothelial projections. The contribution of SKCa and IKCa channels varies between species as well as in different vascular beds of the same species [857].
Table 9.2
Ion carriers of the myoendothelial microdomain between an endothelial cell (EC) and adjoining smooth myocyte (SMC; source: [858]). Signaling in the myoendothelial microdomain through gap junctions and calcium-activated potassium channel enables endothelium-dependent vasodilation. Three types of nanodomains on myoendothelial microdomains (myoendothelial projections) can be defined: (1) myoendothelial gap junction that facilitates ion transfer; (2) KCa channel residence; and (3) transient receptor potential (TRP) channel (non-selective cation carrier) site (TRPC3: type-3 TRP canonical; TRPV4: type-4 TRP vanilloid). The 2 latter nanodomains can merge.
Gap junction (myoendothelial feedback) | |
|---|---|
Molecular transfer | Ions, IP3 |
Endothelial projection membrane | |
KCa3.1 (IK) | K + export (from EC cytosol) |
TRPC3 | Ca2 + import (into EC cytosol) |
TRPV4 | Ca2 + import |
Endothelial projection endoplasmic reticulum membrane | |
IP3R | Ca2 + release |
Smooth myocyte membrane | |
Na + –K + ATPase | K + entry (into SMC cytosol) |
Na + efflux (from SMC cytosol) | |
KIR | K + influx |
The opening of endothelialCa2 + -sensitive KCa3.1 channels on myoendothelial projections elevates the extracellular K + concentration in the myoendothelial space that activatesinwardly rectifying K + channels, which may lodge exclusively on the endothelial surface (in rat mesenteric artery), andNa + –K + pumps on smooth myocytes adjacent to myoendothelial projections, at least in small resistive arteries, thereby hyperpolarizing these cells [859].
In fact, endothelium-dependent vasodilation, at least in human mesenteric arteries, is primarily mediated by [857]: (1) nitric oxide; (2) NO- and PGi2-independent endothelium-derived hyperpolarizing current through IKCa channel; and (3) NO- and PGi2-independent material transfer through gap junction connexin-37 (Table 9.3).
Table 9.3
Features of myoendothelial microdomains in human mesenteric arteries (Source: [857]; MEGJ: myoendothelial gap junction).
Number of SMC layers | ∼ 7 |
|---|---|
Adventitial thickness | 10.8 ± 1.1 μm |
MEGJ density | 1.9 ± 0.7 ×103 μm2 |
KCa3.1 | 4.1 ± 0.6 ×103 μm2 |
Connexin-37 | 2.2 ± 0.5 ×103 μm2 |
An increase in cytosolic Ca2 + concentration in endothelial cells serves as avasodilatory signal, whereas, in smooth myocytes, it triggersvasoconstriction by targeting the actin–myosin stress fibers.
In smooth myocytes, elementary calcium release (calcium sparks) from clusters ofryanodine receptors of the endoplasmic reticulum closely juxtaposed to the plasma membrane activate calcium-sensitivelarge-conductance KCa1.1 channels, thereby causing a transient hyperpolarization that reduces vasoconstriction.
Calcium ion can also be locally released through inositol trisphosphatereceptors of endothelial endoplasmic reticulum to create the so-called endothelialcalcium pulsars in myoendothelial junctions to transmit vasoregulatory signals [860]. Calcium pulsars that encode signals between vascular endothelial cells and smooth myocytes differ from Ca2 + sparks generated by ryanodine receptors. One target of calcium pulsars is KCa3.1 channels in endothelial projections torelax adjoining smooth myocytes.
An intercellular functional unit can thus be defined that is composed of: (1) connexins that form gap junctions; (2) endothelial IP3Rs and KCa3.1 channels; and (3) inward rectifier KIR2.1 channels andvoltage-dependent CaV1.2 channels in smooth myocytes (Fig. 9.1).
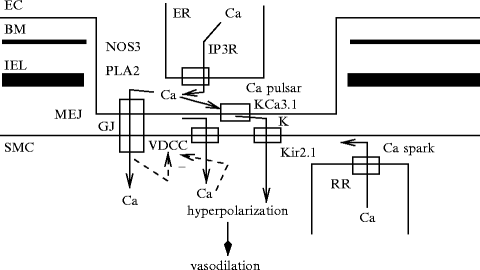
Fig. 9.1
Intercellular functional unit of the myoendothelial junction (MEJ; Source: [860]). Myoendothelial junction is an endothelial domain that crosses the basement menbrane (BM) and internal elastic lamina (IEL) to reach adjacent smooth myocyte membrane. The intercellular functional unit is composed of: (1) connexins that form gap junctions (GJ); (2) inositol trisphosphate receptors (IP3R) and calcium-sensitive KCa3.1 channels (IK) in endothelial cell (EC); and (3) inward rectifier potassium channels (KIR2.1) and voltage-dependent calcium channels (VDCC; CaV1.2) in smooth myocytes (SMC). Calcium pulsar is an endothelial Ca2 + signal that has a restricted localization to MEJ. Its activity is regulated by biological and mechanical agents. Other endothelial calcium-dependent enzymes (e.g., endothelial nitric oxide synthase [NOS3] and phospholipase-A2 [PLA2]) can be activated by calcium pulsars or waves. In smooth myocytes, calcium sparks from clusters of ryanodine receptors of the endoplasmic reticulum closely juxtaposed to the plasma membrane activate Ca2 + -sensitive, large-conductance KCa1.1 channels, thereby causing a transient hyperpolarization that promotes vasodilation.
Two types of Ca2 + signals can cause the opening of IKCa channels in myoendothelial projections [858]. Opening of TRPV4 channels on endothelial membrane causes localized Ca2 + influxes —Ca2 + sparklets — that activate IKCa and SKCa channels. This mechanism ensures response to acetylcholine, at least in mouse small mesenteric arteries. On the other hand,Ca2 + pulsars caused by the opening of clusters of IP3 receptors on parts of the endoplasmic reticulum within the myoendothelial projections may be generated mainly in these microdomains. Calcium pulsars can be triggered by IP3 entering myoendothelial projections from either the endothelial or smooth myocyte cytosol. The former contributes to endothelium-dependent hyperpolarization; the latter to themyoendothelial feedback, by which smooth myocyte contraction is autolimited via K + efflux from the myoendothelial projection, i.e., by which stimulation of vascular smooth myocytes activates endothelial cells to repress vasoconstriction.
In addition,calcium wavelets, another IP3-dependent endothelial Ca2 + signal, play an important role in myoendothelial feedback [858].
Several types of transient receptor potential channels may be involved in calcium entry and myoendothelial signaling: in rat cerebral artery, type-1 TRP ankyrin (TRPA1) and type-3 and -4 TRP vanilloid (TRPV3 and TRPV4) channels; in rat carotid artery, endothelial TRPV4 channel can trigger NO-dependent relaxation as well as, in rat gracilis muscle arterioles and in mouse mesenteric artery, both NO- and EDH-mediated dilation.
Myoendothelial projections are capable of generating localized Ca2 + pulsars vianon-selective cation channel TRPC3, which lodges on the endothelial plasma membrane, in close proximity to both IP3R on the sarcoplasmic reticulum and IKCa on the plasma membrane as well as gap junction, but not on that of vascular smooth myocytes, at least in rat mesenteric artery [861].16 This channel mainly localizes to myoendothelial projections. Protein TRPC3 is distributed throughout the endothelium, but with approximately 5-fold higher density at myoendothelial contact sites. The KCa-mediated endothelial-based vasodilation relies on TRPC3 channels [861].
9.1.3 Vascular Permeability
Angiogenic activity of VEGFa is mainly exerted by upregulating expression oftesticular receptor TR3, or NR4a1 nuclear receptor, in vascular endothelial cells. Transcription factor NR4a1 contributes to the regulation of vascular permeability in 3 contexts [862]: (1) basal vascular permeability that suffices to bring nutrients to cells; (2) acute vascular hyperpermeability, especially in postcapillary venules, in response to short-term exposure to vascular permeabilizing agents such as VEGFa; and (3) chronic vascular hyperpermeability in pathological angiogenesis.
Production of NR4a1 is regulated not only by VEGFa, but also by small vascular permeabilizing agents, such as histamine, platelet-activating factor, and serotonin. Unlike VEGFa that connects toVEGFR2 receptor protein Tyr kinase, histamine, serotonin, and PAF tether to their cognate G-protein-coupled receptors. Like VEGF, NR4a1 acts, at least partly, by increasing indirectly the NOS3 synthesis and decreasing indirectly that of several endothelial cell junction constituents such as cadherin-5.
9.2 Endothelium Types
9.2.1 High Endothelial Venules
High endothelial venules are specialized postcapillary venules of lymphoid tissues, such as lymph nodes and intestine-associated Peyer’s patches [863]. These venules serve as entry of blood-convected lymphocytes into lymphoid organs. Lymphocytes indeed migrate across high endothelial venules for immune surveillance.
High endothelial venules are lined by quasi-cuboidal (plump) endothelial cells rather than flat, thin (except in the nucleus region) ones. These endothelial cells allow lymphocyte extravasation into tissues, using concerted action of integrins, selectins, and chemokines. Lymphocyte capture is initiated by L-selectin and α4β7-integrin. Binding of CCL21 chemokine to its CCR7 receptor activates αLβ2-integrin that mediates lymphocyte arrest and α4β1-integrins. Intercellular adhesion molecule-1 and CCL21 chemokine are upregulated during fever [864].
Member ENPP2 of the ectonucleotide pyrophosphatase/phosphodiesterase family, or autotaxin (Atx), is a secreted enzyme with lysophospholipase-D activity. It converts lysophosphatidylcholine intolysophophatidic acid. It is released by endothelial cells of high endothelial venules. It facilitates the entry of lymphocytes into secondary lymphoid organs. After chemokine activation, naive lymphocytes that search for antigens exit blood stream to lymph node by binding to autotaxin via activated α4β1-integrin on lymphocytes. Extracellular autotaxin produces lysophophatidic acid that enhances lymphocyte motility by binding to cognate G-protein-coupled receptors. Lysophophatidic acid stimulates actin polymerization in primary lymphocytes to trigger their motility [865].
In the spleen, lymphocytes exit blood stream through terminal arterioles that open into the marginal sinus of the spleen. Autotaxin is strongly expressed in central arterioles and spleen marginal zones.
Autotaxin also synthesizessphingosine 1-phosphate that control lymphocyte egress from various lymphoid tissues.
9.2.2 Lymphatic Endothelium
The lymphatic system has many functions. It conveys immunocytes. It drains fluids from the interstitial spaces. It transports absorbed dietary lipids to metabolism sites. Lymphatic vessels develop from specialized venous endothelial cells. During embryogenesis, subpopulations of venous endothelial cells form lymphatic sacs in the region of the primitive subclavian, inferior vena cava, and iliac veins. These sacs then divide to create lymphatic networks. Mediators of embryonic lymphatic development include the transcription factorProspero-related protein-1 (Prox1) and VEGFc. Separation of lymphatic and blood microvasculature in the intestinal mucosa continues beyond fetal life.
Fasting-induced adipose factor (FIAF),17 produced by enterocytes of the small intestine, is required for separation between postnatal intestinal lymphatic and blood vessels [866]. Signaling by FIAF implicates Prox1 effector in the postnatal intestinal lymphatic endothelium. However, lymphaticovenous partitioning also uses Prox1-independent pathways.
9.2.3 Endothelial Fenestrae
Fenestrae (caliber 60–70 nm) exist in thecapillary endothelium, where large molecule exchanges occur between flowing blood and perfused tissues. Fenestrae hence increase the endothelium permeability for water, electrolytes, and small macromolecules, especially in the nephron glomerulus, gastrointestinal tract, liver sinusoids, ocular choriocapillaris, and endocrine glands.
Fenestrae form an array characterized by regular spacing, the so-called sieve plate. The fenestra density in sieve plates can reach about 30 fenestrae per μm2. The fenestra pore is made of 5- to 6-nm openings delineated by a diaphragm with radial fibrils from a central node. Fenestrae are composed of the diaphragm protein PV1, which is required for fenestra formation, as well asactin-filament remodeling [867].
9.3 Endothelial Progenitor Cells
The uninterrupted endothelial lining is maintained and regenerated by both proliferation of endothelial cells and migration of (blood) circulating cells and undifferentiated cells from the subendothelial space. In particular, some blood mononuclear CD34 + cells can acquire endothelial-like characteristics and can home to angiogenesis sites [868].
Circulating endothelial progenitor cells can be incorporated into ischemic tissues as well as at the border of infarcted regions and facilitate neovascularization, as they secrete paracrine factors [868]. They release growth factors and chemokines that stimulate endothelial regeneration by resident endothelial cells. They also contribute to the endothelial lining of microvessels during wound healing.
Circulating endothelial progenitor cells may be involved in re-endothelialization after mechanical vascular injury that preventsintimal hyperplasia. Mobilization of endothelial progenitor cells EPCs may contribute to endothelial regeneration promoted by estrogen, exercise, heme oxygenase-1, and statins [868]. However, homing of circulating endothelial progenitor cells is a minor factor with respect to endothelium growth from the edges of the injured region.
Mobilization, circulation, homing, and local differentiation of bone marrow-derived leukocytes intervene in evolution of most arterial diseases characterized by inflammation.
In adults, the name “endothelial progenitor cell” applied to different cell types. Early endothelial progenitor cells, or endothelial-like cells, have a myelomonocytic origin. They have paracrine effects in neovascularization in vivo. Late endothelial progenitor cells, blood-outgrowth endothelial cells, or endothelial colony forming cells, are highly proliferative. They also participate in neovascularization. Few circulating late endothelial progenitors are CD31 + , CD34 + , CD146 + , PTPRc − , prominin-1 − 18 cells similar to mature circulating and resident endothelial cells [868].
Endothelial progenitor cells orangioblasts can be isolated from blood [869]. Circulating endothelial progenitor cells (CEPC) differ from mature circulating endothelial cells (CEC) by their markers (Table 9.4). Endothelial progenitors and hematopoietic stem cells share numerous surface markers, but the former also expressVEGFR2 receptor.
Table 9.4
Identification markers of endothelial progenitor (EPC) and circulating endothelial cells (CEC; Source: [844]; MCAM: melanoma cell adhesion molecule [a.k.a. cell-surface glycoprotein MUC18 and CD146]; PTPRc: protein Tyr phosphatase, receptor type C [a.k.a. CD45]; SCA1: stem cell antigen-1 [a.k.a. lymphocyte antigen-6A]; SCFR: stem cell factor receptor; vWF: von Willenbrand factor). CD14 is a pattern-recognition receptor. CD133 is the prototypic member of pentaspan transmembrane glycoproteins. CD45 dim is a marker of endothelial progenitor cells. During maturation, the CD45 hematopoietic marker disappears to be replaced by endothelial labels.
Marker | EPC | CEC |
|---|---|---|
Stem cell markers | ||
CD34 | 2 + | ± |
CD133 | 2 + | − |
SCFR | + | − |
Ataxin-1 | + | − |
Endothelial markers | ||
VEGFR2 | 2 + | + |
PECAM1 | + | 2 + |
vWF | + | 2 + |
MCAM |  | 2 + |
NOS3 | + | + |
Leukocyte markers | ||
PTPRc | dim/- | − |
CD14 | 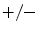 | − |
Bone marrow is a reservoir of stem cells that can regenerate the bone marrow as well as other tissues. Bone marrow is constituted by different types of stem and progenitor cells, such as multipotent adult progenitor cells that can, at least in vitro, generate many cell types as well as mesenchymal stem cells, and hemangioblasts. However, the contribution of bone marrow-derived progenitor cells to repair of vascular damages is rather limited.
9.3.1 Hemangioblast
Hemangioblast is a common precursor for hematopoietic and endothelial cells under the influence of growth factors. Hematopoietic stem cells give birth to lymphoid and myeloid progenitor cells;19 vascular stem cells are precursors of endothelial progenitors and, secondarily, endothelial cells and pericytes.
Hemangioblasts are mobilized by several factors mainly via activatedendothelial nitric oxide synthase andmatrix metallopeptidase MMP9 produced in bone-marrow niches [870]. Nitric oxide availability rises on signaling bygrowth hormone andinsulin-like growth factor-1. On the other hand, NO inhibitors (e.g., asymmetric dimethylarginine [ADMA]) prevent mobilization and differentiation of endothelial progenitor cells, their incorporation into endothelial tube-like structures, and formation of colony-forming units from cultured peripheral blood mononuclear cells [870].Estrogens mobilize endothelial progenitors via estrogen receptor ERα and ERβ (a.k.a. nuclear receptors NR3a1 and NR3a2), MMP9, and NOS3 [870].Smoking increases oxidative stress and reduces NO availability.
Hemogenic endothelial cells that line blood vessels in the embryo give rise to progenitors of blood cells in fetal liver and adult bone marrow. Hematopoietic progenitor cells can actually form clusters attached to the endothelium of the ventral wall of the abdominal aorta during embryogenesis.
9.3.2 Endothelial Progenitor Cell – Circulating Angiogenic Cell
Endothelial progenitor cells, orcirculating angiogenic cells,20 can reside in the bone marrow and blood as well as adventitia and endothelium [868].
Endothelial progenitor cells are activated by stimuli for tissue regeneration, such asvascular endothelial growth factor,placental growth factor,granulocyte–monocyte colony-stimulating factor (CSF2),granulocyte colony-stimulating factor (CSF3),erythropoietin,21angiopoietin-1, and CXCL12 chemokine, are recruited from the bone marrow into blood flow to be convected toward angiogenesis sites (Table 9.5) [870, 871].22 Circulating endothelial progenitor cells yield protection by their innate ability to replace dysfunctional or damaged endothelium.
Factor | Effect |
|---|---|
Age | 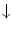 EPC mobilization, survival, activity EPC mobilization, survival, activity |
Estrogen | 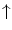 EPC concentration EPC concentration |
Exercise | 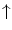 EPC concentration EPC concentration |
Growth factors |  EPC mobilization EPC mobilization |
Hypercholesterolemia |  EPC proliferation, migration, survival EPC proliferation, migration, survival |
Hypertension |  EPC proliferation EPC proliferation |
 EPC survival EPC survival | |
Smoking | 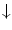 EPC density EPC density |
However, during angiogenesis, slight recruitment of bone marrow-derived cells, in particular VEGFR2 + precursors, does not contribute to vascular endothelium [872]. Bone marrow-derived cells that express platelet–endothelial cell adhesion molecule PECAM1, VEGFR1, and VEGFR2 are always stromal or perivascular cells. Perivascular hematopoietic cell populations that can produce endothelial markers are not bone marrow-derived endothelial cells.
9.3.2.1 Early and Late Endothelial Progenitors
Two populations of endothelial progenitor cells exist — early and late endothelial progenitors — with distinct growth patterns and secretion modalities of angiogenic factors. Spindle-shaped early endothelial progenitors are also called early-outgrowth endothelial progenitors, early-outgrowth culture-expanded endothelial progenitor cells, endothelial cell-like cells, colony-forming unit (CFU) of endothelial cells, circulating angiogenic cells, attaching cells, and culture-modified mononuclear cells [870].
In fact, this population of endothelial progenitor cells can be subdivided into 2 classes. The first reported progenitor cell type — the colony-forming unit-Hill cells — originates from cultures of non-adherent peripheral blood mononuclear cells that are not able to form vascular structures in vivo [873]. Circulating angiogenic cells — early-outgrowth cells — are descendants of the monocyte–macrophage subset and operate in initiation of angiogenesis during wound healing and tissue remodeling [873]. Early-outgrowth endothelial progenitors express VEGFR2, PECAM1, cadherin-5, CD34 (generally at a low level), and von Willebrand factor, as well as monocytic marker CD14 (bacterial lipopolysaccharide receptor component) and panleukocytic marker PTPRc [870].23
Late endothelial progenitor cells are also named late-outgrowth endothelial progenitors, endothelial colony-forming cells (ECFC), and blood-derived outgrowth endothelial cells. They start to proliferate only after 2 to 3 weeks in culture [870], but possess a high proliferative capacity [873]. These cells can spontaneously form blood vessels. They express all typical properties of endothelial cells. They express endothelial markers, like VEGFR2, melanoma cell adhesion molecule (MCAM or CD146), and cadherin-5, in addition to CD34, but not hematopoietic markers, such as PTPRc and monocyte differentiation antigen CD14 [870].
Circulating endothelial progenitor cells that include cells that generate both early- and late-outgrowth endothelial progenitors may be represented by CD34 + , VEGFR2 + cells [870].
Hemangioblasts do not possess receptor protein Tyr phosphatase PTPRc (or CD45), a common leukocyte antigen. Circulating endothelial progenitors are defined by markers, such as VEGFR2, hematopoietic progenitor cell glycoprotein and intercellular adhesion factor CD34, PTPRc, and pentaspan transmembrane glycoprotein prominin-1.24 Monocytes can also provide a source of endothelial progenitors that do not proliferate, but release angiogenic growth factors.
9.4 Endothelial Cell Migration
Endothelial cells migrate during angiogenesis (Chap. 10; Vol. 2 – Chap. 6. Cell Motility).Chemokines of the CXC set enhance the migratory capacity of endothelial cells and facilitate homing of endothelial progenitor cells into ischemic tissues. Chemokine ligand CXCL1225 promotes tubulogenesis of microvascular endothelial cells via enhanced expression of growth factorsVEGF andFGF2.Hypoxia-inducible factor HIF1 primes CXCL12 synthesis in ischemic tissue for cell recruitment and homing, especially of vascular endothelial cells and circulating progenitor cells that express chemokine CXCR4 receptor,26 thereby boosting tissue regeneration.
Chemokine CXCL12 activates the PKB–NOS3 axis as well asmitogen-activated protein kinases, such asERK1 and ERK2, JNK, and P38MAPK, in different cell types. Activatednitric oxide synthase NOS3 then produces nitric oxide that subsequently nitrosylates (inactivates)mitogen-activated protein kinase phosphatase MKP7. Phosphatase MKP7 then cannot inhibit Jun N-terminal kinase JNK3 that binds to its adaptor β-arrestin-2 [874].27 In bovine aortic endothelial cells, CXCL12 activates an NOS3-independent pathway that targets ERK1 and ERK2 to initiate cell migration.
9.5 Molecular Expression in the Vascular Endothelium
The vascular endothelium synthesizes numerous molecules to regulate its behavior and to respond to environmental cues, as well as to control its surrounding (Table 9.6, Fig. 9.2).
Table 9.6
Examples of endothelial cell production. Many manufactured molecules are stored and released upon calcium stimulation. All substances stored in granules (e.g., vWF, tPA, TFPI, protein-S, and ET1 and ET3) are released by regulated exocytosis. Certain substances such as tissue plasminogen activator (tPA) are constitutively released, but acute exocytosis from small tPA-containing vesicles and caveolae is primed by compounds, such as histamine, endothelin (ET), and cytokines. Endothelial cells control hemostasis, as they produce both urokinase-like (uPA) and tissue-type plasminogen activators for fibrinolysis as well as plasmalemmal thrombomodulin (TM that links to thrombin and cleaves carboxypeptidase-B2, or thrombin-activable fibrinolysis inhibitor [TAFI], into its active form, in addition to be a cofactor that enhances the thrombin-catalyzed activation of anticoagulant protein-C), and antithrombin-3 (a proteolytic inhibitor), in addition to secretion of a specific thrombin receptor and peptidase nexin (an inhibitor that binds to thrombin and plasminogen activators). Endothelial cells also secrete matrix constituents, such as fibronectin (FN), various types of collagens, laminin (Ln), tropoelastin, von Willebrand protein (vWF), and thrombospondin (Tsp).
Structure | Proteoglycans (hyaluronic acid, DS, HS, KS) |
|---|---|
Glycoproteins (Ln, FN, Tsp, vWF) | |
Collagens-4/8, | |
Survival | HETE |
Growth | Growth factors (PGDF, gmCSF, gCSF, mCSF) |
Growth mediators (NO, 5HT, PGI2, TXA2, Ang2, ROS, TNFα) | |
Hormones (CNP) | |
Motility | Chemokines (Lkt, HETE) |
Semaphorins, plexins | |
Guidance | Delta and Notch, EPH and ephrins, netrin and Unc, |
Slit2 and Robo4 | |
Vasomotor | ET, NO, H2S, PGH2, PGI2, TXA2, EDRF, HETE, EET, |
KLF2, adrenomedullin, ATn2, 5HT, ACh, ATP, | |
ACE, ROS, CNP | |
Adhesion | Integrin, selectin, cadherin, |
ICAM1, ELAM1, | |
13HODE, TxnIP | |
Inflammation | His, Bdk, Ang2, NO, ROS, TNFα |
Coagulation | PAI1, PGI2, TM, tPA, uPA, TFPI, NO, Protein S, |
TXA2, vWF, FV, FIII, PAI, | |
heparin sulfates, ectonucleotidases | |
Hormone | Adiponectin, CNP, |
prolactin, growth hormone, placental lactogen |
Fig. 9.2
Effects of the endothelial cell on its environment (Source: [875]). (1) Endothelial cells regulate endothelial permeability for plasma substances and adhesion of blood cells (promigration substances: selectins, IgCAMs; antimigration molecules: nitric oxide). They sequester leukocyte-interactive proteins, such as P-selectin and chemokines. They repress the synthesis of certain adhesion molecules, such as E-selectin, vascular cell adhesion molecule VCAM1 and, intercellular adhesion molecule ICAM1. (2) Endothelial cells release clotting factors and anticoagulation agents, such as nitric oxide (NO), prostacyclin (PGI2), tissue factor pathway inhibitor (TFPI), tissue plasminogen activator (tPA), and thrombomodulin (TM). Agent TFPI prevents the initiation of coagulation, as it inhibits factor VIIa–tissue factor complex. Heparan sulfate proteoglycans bind anti-thrombin-3 to inactivate thrombin. Thrombomodulin binds thrombin and diverts its activation activity from fibrinogen to protein-C that, in coordination with protein-S, inactivates several clotting components. Both NO and PGI2 synergistically impede platelet adhesion and aggregation. (3) Endothelial cells produce growth regulators, either progrowth, such as angiotensin-2 (ATn2), platelet-derived growth factor (PDGF), colony-stimulating factor (CSF), and endothelin (ET), or antigrowth molecules, such as NO and transforming growth factor-β(TGF), especially for smooth myocytes. (4) Endothelial cells synthesize vasoactive substances.
Vascular endothelial cells, like vascular smooth myocytes, possess enzymes of the cytochrome-P450 superfamily, such as epoxygenases and ω-hydroxylases that metabolize arachidonic acid released from phospholipids of the plasma membrane by activated phospholipase-A2 into vasoactive substances.
20-Hydroxyeicosatetraenoic acid (20HETE) is produced bycytochrome-P450 in vascular smooth myocytes as well as uniquely in endothelial cells of pulmonary arteries. Compound 20HETE enhances reactive oxygen species production by pulmonary arterial endothelial cells using NADPH oxidase and promotes angiogenesis. Agent 20HETE activates NADPH oxidase. It protects from apoptosis, as it activatesphosphatidylinositol 3-kinase [876].
In human dermal microvascular endothelial cells, 20HETE provokes a rapid and sustained increase in superoxide synthesis by NADPH oxidase. It also raises the production ofvascular endothelial growth factor that, in turn, upregulateshypoxia-inducible factor HIF1α via the ERK1/2 pathway [877]. Moreover, it heightens the expression of erythropoietin receptor and angiopoietin-2 via HIF1α.
Products of cytochrome-P450 epoxygenase and ω-hydroxylase as well as reactive oxygen species derived from NADPH are intracellular signal transducers for proliferation of vascular cells (via extracellular-regulated protein kinases ERK1 and ERK2) as well as angiogenesis.
In vascular endothelial cells subjected to acetylcholine, bradykinin, or shear stress, activated phospholipases producearachidonic acid that is processed by cyclooxygenases, cytochrome-P450s, and lipoxygenases. In endothelium of some arteries, a substantial component of vasodilation depends onlipoxygenase-induced arachidonic acid metabolites. Arachidonate 15-lipoxygenase (ALOx15) synthesizes vasoactive metabolites such as15-hydroxy (11,12)-epoxyeicosatrienoic acid that is hydrolyzed by soluble epoxide hydrolase to(11,12,15)-trihydroxyeicosatrienoic acid. Hydroxyepoxy- and trihydroxyeicosatrienoic acids areendothelium-derived hyperpolarizing factors that activatecalcium-activated, small-conductance KCa2 channels [825]. In vascular endothelium of other arteries, arachidonate 12-lipoxygenase (ALOx12) produces12-hydroxyeicosatetraenoic acid that relaxes smooth myocytes via calcium-activated, large-conductance KCa1 channel [825].
Insulin activates the PI3K pathway not only to stimulate glucose uptake, but also to promote synthesis ofnitric oxide in the endothelium. Insulin hence favors endothelium-dependent relaxation of vascular smooth myocytes (Sect. 9.10).
9.5.1 Caveolae
Caveolae are specialized, invaginated membrane rafts, i.e., dynamic assemblies of sphingolipids and cholesterol, that contribute to vesicular transport and signaling. They indeed concentrate or segregate receptors and signaling intermediates. These nanodomains constitute platforms on which kinases and phosphatases can operate. Caveola-mediatedendocytosis differs from other types of endocytosis by chemical sensitivities, cargos, adaptors, and signaling proteins (Vol. 1 – Chap. 9. Intracellular Transport).
The organization and function of caveolae depend on coat caveolins and adaptor cavins (cavin-1–cavin-4) that promote membrane remodeling and caveolin-derived structure transfer. Caveolae can form transendothelial channels and vesiculovacuolar organelles and cavicles.
Caveolin-1 and -2 lodge in most cell types of the cardiovascular apparatus, whereas Cav3 is expressed primarily in myocytes (cardiac, skeletal, and smooth myocytes). Caveolin-1 or -3 is needed for the formation of caveolae, but not Cav2. Caveolin-1 resides in the plasma membrane andGolgi body. Exocytosis from the Golgi body to the plasma membrane is regulated by amyloid-β protein and insulin [878]. In addition, Cav1 endocytosis is controlled byNa + –K + ATPase. Caveolin-2 supports caveola assembly via its hetero-oligomerization with Cav1.
9.5.1.1 Regulators of Caveolae
Cavins (cavin-1–cavin-4) regulate caveolin density as well as caveola morphology [878].28 Cavins possess leucine zipper-like domains for between-protein interactions, PEST domains for protein turnover, and phosphorylation motifs. They bind phosphatidylserine. They are phosphorylated upon insulin stimulation [878].
Cavin-1 colocalizes with Cav1 in membrane rafts, where they interact with the cell cytoskeleton, especially cortical microtubules and actin filaments [878]. Cavin-1 sequesters caveolins into caveolae. Cavin-2 binds to and recruits cavin-1 to the plasma membrane. The cavin-1–cavin-2 complex stabilizes Cav1-containing structures. Cavin-2 is a PKC substrate that is involved in PKC compartmentation in caveolae. Cavin-3 supports budding and formation of cavicles. Cavin-4 is predominantly in myocytes.
Dynamin-2 binds to Cav1 during Cav1-mediated endocytosis.Insulin receptor as well asSrc kinase phosphorylate Cav1 (Tyr14), in response to growth-factor stimulation and cellular stress. Caveolin-1P interacts withC-terminal Src kinase to preclude Src action [878].
9.5.1.2 Caveolae in Endothelium Functions
Caveolin-1 regulates microvascular permeability, Ca2 + influx, vascular remodeling, and angiogenesis. Many G-protein-coupled receptors, receptor and cytosolic Tyr kinases (e.g., EGFR), small GTPases, and components of the MAPK module (e.g., Raf, ERK1, and ERK2) interact with Cav1 and reside in caveolae [878]. In particular, caveolin-1 sequesters ERK1, and ERK2, thereby impeding the activity of this pathway.
In addition, caveolin-1 contributes tointegrin signaling, particularly β1-integrin localization to caveolae upon IGF stimulation. Caveolin-1 also tethers cyclo-oxygenase-2 to the endoplasmic reticulum, hence promoting its degradation.
In vascular endothelial cells, caveolae participate in the regulation of the vascular tone, as caveolin-1 in caveolae inhibits endothelial nitricoxide synthase (NOS3) activity, but not caveolin-1 in non-caveolar membrane rafts [878]. Caveolin-1 and Ca2 + ions antagonistically regulate NOS3 in the microcirculation. However, caveolin-1 participates in Ca2 + import into endothelial cells.29
9.5.2 Nuclear Receptors
Drug catabolism ensures the body’s protection against toxics. The nuclear receptor NR1i230 controls drug clearance via transcription of genes involved in drug transport (multidrug resistance transporter MDR1) and metabolism (conjugation [glutathione transferase] and oxidation [cytochrome-P450 CyP2b, CyP2c, and CyP3a, and glutathione peroxidase]). In human vascular endothelial and smooth muscle cells, activated NR1i2 stimulates expression of the Mdr1, CYP3A, CYP2B, and CYP2C genes as well as increases cellular level of glutathione and activity of glutathione peroxidase to protect the vasculature, in particular against oxidative stress [879].
9.5.3 Examples of Endothelial Receptors
9.5.3.1 Endothelial Protein-C Coreceptor
Endothelial cells express endothelial protein-C receptor (EPCR) to regulate theprotein-C anticoagulant and anti-inflammatory pathway via thethrombin–thrombo modulin complex. Cytoprotective activated protein-C can also upregulate anti-apoptotic and anti-inflammatory gene expression.
Vascular smooth myocytes also express EPCR [880]. In smooth myocytes, activated protein-C induces phosphorylation of extracellular signal-regulated kinases ERK1 and ERK2 viapeptidase-activated PAR1 receptor. Effect of activated protein-C is significantly enhanced in the presence of thrombin.
However, thrombin does not engage EPCR coreceptor; it cleaves completely PAR1 that is subsequently internalized and degraded [881]. Thrombin binds to PAR1 and activates preferentially subunits of the Gq and G12/13 subclasses of G protein heterotrimer to initiate Ca2 + mobilization and PKC activation on the one hand and to disrupt the endothelial barrier using monomeric RhoA GTase, on the other.
On the other hand, activated protein-C connects to EPCR and causes a limited cleavage of PAR1 receptor [881]. Moreover, activated protein-C and thrombin stimulate Rac1 and RhoA GTPases using G protein and β-arrestin, respectively [882]. Therefore, activated protein-C operates as a biased agonist that activate β-arrestin signaling.31 Activated protein-C releases β-arrestin-2 from PAR1, which then interacts withDisheveled-2, a scaffold and mediator of the Wnt–Frizzled signaling, which polymerizes and protect the endothelial barrier.32
9.5.3.2 Peptidase-Activated Receptors
Peptidase-activated receptor PAR1 on endothelial cells contributes to cell responses that trigger or prevent blood coagulation and ensures cell protection [882]. Both thrombin and activated protein-C stimulate PAR1, but cause opposite effects.
Serine peptidase thrombin binds to PAR1 and cleaves its extracellular domain to form a tethered ligand to activate PAR1-mediated inflammation and increase endothelial barrier permeability.
The anticoagulant peptidase activated protein-C stimulates a subpopulation of PAR1 that colocalizes with their coreceptor, endothelial protein-C receptor, PAR1, in membrane nanodomains enriched in caveolin, to promote endothelial barrier protection (Table 9.7). Receptor PAR1 localizes to caveolae connected to β-arrestin in unstimulated cells. The PAR1–EPCR couple supports cytoprotection. Activated protein-C recruits and activates Disheveled-2 [882].
Table 9.7
Activation by thrombin and APC of peptidase-activated receptor PAR1 and opposite effects (Source: [882]; Arr: arrestin; Dvl: Disheveled; EPCR: endothelial protein-C receptor).
Messenger | Effect | Pathway |
|---|---|---|
Thrombin | Endothelial barrier | PAR1–Gαq–Ca2 + –PKC |
disruption | PAR1–Gα12 ∕ 13–RhoA | |
Activated | Endothelial barrier | PAR1–EPCR–Cav–βArr– |
protein-C | maintenance | –Dvl2–Rac1 |
Hypoxia primes an angiogenic phenotype in endothelial cells.Hypoxic cancer cells upregulates in endothelial cells protease-activated receptor PAR2 and pro-angiogenic heparin-binding EGF-like growth factor(HBEGF) and increases phosphorylation ofERK1 and ERK2 [883]. Tissue factor that triggers PAR signaling is induced by hypoxia in several types of cancer cells; however, tissue factor remains undetectable in hypoxic endothelial cells, although several stimuli (e.g., shear stress and growth factors) transiently cause induce tissue factor production TF in endothelial cells.
9.5.3.3 I-Peptide Receptor
Endothelial cells express carbohydrate I-peptide receptor (IPR) that is responsible for lung colonization ofcancer cells [884]. Receptor IPR corresponds to alternatively spliced variants of Arg/Ser-rich splicing factors (SFRS1, SFRS2, SFRS5, and SFRS7). Like many carbohydrate-binding proteins of the C-type lectin family, SFRS protein that is not a C-type lectin requires calcium to bind to carbohydrates, especially fucosylated oligosaccharides.
9.5.3.4 FGFRs
Fibroblast growth factor FGF2 provokes endothelial cell migration and angiogenesis via 2 types of receptors: high-affinity receptor protein Tyr kinases such asFGFR1 and heparan sulfate proteoglycans such as transmembranesyndecan-4, a FGFR1 coreceptor. The latter determines the kinetics and magnitude of FGF2-inducedMAPK signaling (ERK1 and ERK2) by promoting the macropinocytosis of the FGFR1–syndecan-4–FGF2 complex using RhoG and Rab5 GTPases [885]. Small RhoG GTPase promotes membrane ruffling and macropinocytosis. Monomeric Rab5 GTPase is involved in early signaling endosomes. Signaling from FGFR1 initiates MAPK activation; syndecan-4-dependent FGFR1 macropinocytosis modulates the kinetics of MAPK activation.
9.5.3.5 VEGFRs
Angiogenic vascular endothelial growth factor VEGFa is also a potent vascular permeabilizing factor, whereas endothelial growth factors, such as FGF2 and PDGF, do not affect vascular permeability.
The VEGFR receptors participate in signaling pathways that control and coordinate transcriptional, post-transcriptional, and post-translational mechanisms involved in the control of endothelial cell behavior duringangiogenic sprouting, branching with endothelial leading tip and trailing stalk cells, and tubulogenesis. Stalk cells support the extension of sprouting vessels, generate the trunk of new vessels, build a vascular lumen in growing vessels, and maintain connection with the parental vessel.
During angiogenesis, signaling launched by pro-angiogenic ligands (i.e., autocrine VEGFa and VEGFc regulators) of VEGFR2 and VEGFR3 select tip cells for sprouting [886]. Angiogenic sprouting is guided by gradients of pro-angiogenic growth factors and various guidance cues, such as semaphorins and ephrins. The navigators Uncoordinated-5 homolog Unc5b (receptor of secretednetrins),Roundabout homolog Robo4,plexin-D1,neuropilins,ephrin-B2, andEPHb4 receptor are major conductors of angiogenesis.
On the other hand, the decoy receptor VEGFR1 limits tip cell formation; Delta-like ligand DLL4 of Notch receptor prevents tip cell fate in endothelial cells adjacent to tip cells [886]. In addition, Roundabout homolog Robo4, and Wnt signaling in stalk cells repress tip cell behavior to maintain the hierarchical organization of sprouting endothelial cells.
Vascular tubulogenesis is initiated by the acquisition of the apicobasal polarity of endothelial cells that is regulated by cell–matrix interactions and signaling via partitioning defective protein PAR3 and VEGFR receptor [886].
9.5.3.6 TGFβ Receptors
Endothelial cells produce receptor protein Ser/Thr kinasesTβR2, TβR1 (or ALK5), and ALK1 activin receptor-like kinase. Unlike inhibition in quiescent endothelium of cadherin-5 on the VEGF–ERK1/2 pathway, cadherin-5 at adherens junctions that is stimulated by TGFβ recruit TβR2 and enhance the assembly of TβR2–TGFβR1 heteromers [887].
Activated TGFβR1 phosphorylates receptor-associated SMAD proteins (rSMAD). Receptors ALK1 and ALK5 phosphorylate SMAD1, SMAD5, and SMAD8 and SMAD2 and SMAD3, respectively. Cadherin-5 is needed for potent, sustained phosphorylation of SMAD1/5 and SMAD2/3 by TβR1 receptor. Phosphorylated cytoplasmic rSMAD dissociates from the receptor and complexes with SMAD4 mediator. The rSMAD–SMAD4 complex accumulates in the nucleus and interacts with specific DNA-binding proteins to regulate transcription. Antiproliferative and antimigratory signaling by TGFβ thus contributes to Cdh5-dependent stabilization and remodeling of the vascular endothelium. Activation by SMAD2 and SMAD3 is indeed stronger than SMAD1 and SMAD/5 stimulation. Consequently, the TGFβ–ALK5–SMAD2/3 pathway is more efficient than TGFβ–ALK1–SMAD1/5/8 signaling that promotes cell growth and motility.
Proliferating endothelial cells synthesize a large amount ofendoglin, a modulator of ALK1 and ALK5, which is bound by TGFβ. Lack in endoglin or ALK1 causes defects in cardiovascular development. In particuler, endothelial cells of the endocardium do not undergo mesenchymal transition required for their migration into the atrioventricular cushion.
Vascular endothelial cells can transform into multipotent stem-like cells using ALK2 receptor [888]. Constitutively active ALK2 (upon mutations of the Alk2 gene) in endothelial cells or exposure to its ligands TGFβ2 or BMP4 causes endothelial-to-mesenchymal transition and acquisition of a stem cell-like phenotype. These mesenchymal stem-like cells can differentiate into osteoblasts, chondrocytes, and adipocytes.33
9.5.3.7 BMPRs
Bone morphogenetic proteins link toBMPR1 and BMPR2, thereby causing phosphorylation of BMPR-specificSMADs (SMAD1, SMAD5, and SMAD8). Blood flow provokes BMP4 synthesis in endothelial cells.
Endothelial cells exposed to time-dependent shear stress have higher turnover (and proliferation) rate than those bearing steady shear stress. The SMAD proteins can modulate the cell cycle, which is controlled by cyclin-dependent protein kinases coupled to their regulatory subunits cyclins and inhibitors CKI1a and CKI1b. In endothelial cells, SMAD1 and SMAD5 can be activated from BMPR in the absence of BMP ligands to promote cell proliferation, unlike BMP4-dependent ICAM1 expression by nuclear factor-κB in the context of inflammation to support monocyte adhesion to the endothelium [889]. Time-dependent shear stress actually causes a sustained activation of SMAD1 and SMAD5 and subsequently ofTOR andS6K (or P70RSK) kinases, thereby upregulating cyclin-A and downregulating CKI1a and CKI1b [889]. Shear-mediated activation of TOR and S6K is modulated by the interplay between AMPK inhibitor and PKB activator.
9.5.3.8 Neurotrophin Tyr Kinase Receptors
Neurotrophins, such as nerve growth factor, brain-derived neurotrophic factor (BDNF), and neurotrophin-3 and theirhigh-affinity neurotrophin Tyr kinase receptors (NTRK2–NTRK3)34 and low-affinity nerve growth factor receptor (NGFR, or TNFRSF16)35 localize to pulmonary vasculature and airways in humans. Both ligands and receptor are produced in the same cell type to create auto-, juxta-, or paracrine effects. In addition, endothelial cells of the systemic circulation produce NTRK receptors. Moreover, BDNF circulates in blood (15–30 ng/ml).
Neurotrophins rapidly trigger (6–8 mn)nitric oxide production in pulmonary endothelial cells [890]. Neurotrophins BDNF and NT3 act predominantly via high-affinity NTRK2 and NTRK3 receptors, respectively, with partial involvement of low-affinity TNFRSF16 receptor. Upon NTRK autophosphorylation, both BDNF and NT3 increase phosphorylation of PKB that, in turn, phosphorylates NOS3 kinase. As, in smooth myocytes, neurotrophins increase intracellular Ca2 + concentration that promotes vasoconstriction, their net effect on vascular tone is determined by the relative contribution of neurotrophins on endothelial cells with NO generation that elicits vasodilation.
9.5.3.9 Angiotensin-2 Receptors
Angiotensin-2 is a vasoconstrictor and progrowth factor that operates viaAT1 receptor. On the other hand, AT2 receptor transmits signals via bradykinin and nitric oxide to cause vasodilation.
9.5.3.10 Roundabout Receptors and Slit Ligands
TheSlit–Robo signaling complex is required for the development of the heart and blood vessels. Endothelial-specific Robo4 maintains vascular integrity [892]. Activation of Robo4 by Slit2 inhibits VEGF-induced migration along angiogenic stimulus gradients and vascular leak.
Slit proteins steer the migration of many cell types once bound to Robo receptors. Signaling incorporates Rho GTPase-activating proteins (RhoGAPs). ProteinRhoGAP39 promotes hydrolysis of RacGTP and, less efficiently, of CDC42GTP [893]. Guided cell migration also intervenes in the development of the respiratory tract. In addition, Rac and possibly CDC42 act downstream of Robo in midline repulsion of brain axons.
9.5.3.11 Cytokine Receptors
Endothelial cells expressIL20 receptors.Interleukin-20 of the interleukin-10 family causes proliferation of endothelial cells, thereby promoting angiogenesis. Interleukin-20 favors phosphorylation of ERK1, ERK2, P38MAPK, and JNK kinases [894].
9.5.4 Ion Carriers of Endothelial Cell
Ion channels abound in the plasma membrane of non-excitable endothelial cells [895]. They control ion influx necessary to produce vasoactive molecules, pro- and anticoagulants, growth factors, etc., that are released for storage or immediate action (Table 9.8). They are involved in the regulation of cell transport. Angiogenesis entails mediation of ion channels.
Table 9.8
Role of endothelial ion channels. Ion channel expression depends on cell culture features and growth conditions, as well as endothelial cell type. Endothelial cells express numerous ion channels that intervene in many processes between endothelial cells and between endothelial cells and smooth myocytes. Two major functions of ion channels are the control of calcium influx and the modulation of membrane potential. The membrane potential provides the electrochemical driving force for Ca2 + influx. In endothelial cells, Ca2 + -dependent K + currents are modulated by αVβ3-integrin attachment to fibronectin. Potassium outflux through KIR channels enhances cell migration.
Function | Examples of produced mediators |
|---|---|
Synthesis of vasoactive substances | NO, ET, PGi2, TxA2 |
Production of hemostatic factors | vWF, PAF, TPFI |
Secretion of fibrinolytic factors | tPA |
Generation of growth factors | EGF, FGF2/4, HGF, IGF1/2, PDGF, VEGF |
Molecular transport | Molecular machinery |
Mechanotransduction | NOS activation |
Cell proliferation | NO |
Cell extravasation | Cell adhesion molecules |
Endothelial cell–matrix adhesion | Cell adhesion molecules |
Endothelial cell migration | Chemokines |
Angiogenesis | Angiomotin, angiopoietin, angiokines, |
growth factors, Notch, DCC, ephrin, EPH, | |
semaphorins, Slit-2/3, SlitR, VEGFR |
Endothelial cells undergo slow and more or less small changes in membrane potential associated with the activity of plasmalemmal ion channnels and pumps. The membrane potential of vascular endothelial cells is negative compared with the blood and tissue. The membrane potential is in general more negative in macrovascular than microvascular endothelial cells [895].37
Ion fluxes between endothelial cells, between endothelial and smooth muscle cells across the basement membrane and myoendothelial junctions, and between endothelial cell and migrating lymphocyte or monocyte, also occur through gap junctions. The large variability in gap junctions depends on the connexin isoforms (Cx37, Cx40, and Cx43) that are expressed in endothelial cells.38 The electrical coupling via high-conductance gap junctions between endothelial cells and smooth myocytes is particularly important in arterioles.
9.5.4.1 Calcium Channels and Calcium Signaling
Calcium influx channels are important for long-lasting responses that are required for the release of various compounds (e.g., nitric oxide, platelet-activating factor, tissue factor pathway inhibitor, tissue plasminogen activator, and von Willebrand factor; Sect. 9.8; Tables 9.9 to 9.11).
Table 9.9
Calcium channels and pumps of endothelial cells (Source: [895]). (Part 1) Non-selective cation channels (NSC). Endothelial cells respond to most stimuli with a calcium influx. The depolarizing action of Ca2 + influx is compensated by activation of Ca2 + -dependent K + channels. In addition, non-selective cation channels, permeable to Ca2 + or not, can generate a negative feedback on calcium influx by membrane depolarization.
Ion channels | Features |
|---|---|
Purinoceptor | ATP and shear stress-induced |
(P2X4) | Ca2 + entry |
Superoxide-gated NSC | Na + , K + , and Ca2 + permeability |
Receptor-activated cation channels | Activation by PLC, IP3, and Ca2 + |
(Ca2 + -permeable; RACC) | (positive feedback) |
(TRP3/4/5/6 ?) | NOS3 stimulation |
Amiloride-sensitive NSC | Blood–brain barrier |
Cyclic nucleotide-activated NSC | Negative feedback on Ca2 + influx |
CGN2 | Activation by the NOS3–NO–cGMP axis |
Hyperpolarization-activated | Blood–brain barrier |
cyclic nucleotide-gated channels | NO- and cGMP-dependent cell processes |
(HCN; Ca2 + permeable) | Modulation by NO, cGMP, and cAMP |
Table 9.11
Calcium channels and pumps of endothelial cells (Source: [895]). (Part 3) Store-operated (SOC) and other channels. Activated VEGFR2 triggers activation of Ca2 + -release-activated Ca2 + -channel (CRAC) channels.
Ion carriers | Features |
|---|---|
Store-operated channels (SOC) | |
Ligand-gated Ca2 + -selective | Activation by IP3, ATP, |
channels | and bradykinin |
CRAC | Smaller Ca2 + selectivity and |
higher conductance than usual CRAC | |
Ryanodine receptors | Ca2 + influx |
Na + –Ca2 + exchanger | Ca2 + entry |
Modulator (Ca2 + efflux) | |
Plasma membrane Ca2 + pump | Modulator (Ca2 + efflux) |
Sarco(endo)plasmic reticulum | Ca2 + efflux |
Ca2 + pump | Modulator of plateau phase |
(SERCA3) | of Ca2 + influx |
Mitochondrial Ca2 + uniporters | Modulator (Ca2 + efflux) |
Mechanosensitive | Na + , K + , and Ca2 + transport |
Ca2 + -permeable | Inhibition by PKG |
channels | Negative feedback by NOS3 activation |
Shear stress-activated NSC | Permeability: Ca2 + > Na + |
Two types ofCa2 + signals include: (1) calcium oscillations and (2) biphasic increase in intracellular calcium level with a fast peak followed by a long-lasting plateau. Fast transient Ca2 + peak is caused by IP3-induced Ca2 + release from intracellular stores, whereas the plateau phase results from Ca2 + influx from the extracellular space activated by store depletion.
Table 9.10
Calcium channels and pumps of endothelial cells (Source: [895]). (Part 2) Transient receptor potential channels (TRP). Channels TRPC1 and TRPC6 are highly expressed by endothelial cells, whereas TRPC3, TRPC4, and TRPC7 are weakly produced. Channels TRPC1, TRPC4, and TRPC5 are store-operated channels, whereas receptor-operated channels TRPC3 and TRPC6 are activated independently of store depletion. Multiple heterologous combinations of TRPCs (e.g., TRPC1–TRPC4, TRPC1–TRPC5, and TRPC3–TRPC6) generate tetrameric channels. Both TRPV1 and TRPV4 are involved in endogenous cannabinoid-dependent vasorelaxation. Channel TRPC4 participates in ATP-primed vasoconstriction and acetylcholine-induced vasorelaxation.
Ion channels | Features |
|---|---|
(TRPC1–TRPC7; TRPV1/2/4; TRPM1–TRPM8 except TRPM5, | |
and TRPP1/2 are expressed in endothelial cells) | |
TRP | Variable expression among EC type |
Ca2 + influx (except TRPM4) | |
TRPP1–TRPP2 complex | Mechanosensing |
TRPC1/6, TRPV2, TRPM4 | Stretch (pressure) activation |
TRPV4 | Shear activation (stretch-insensitive) |
TRPC1–TRPC7 | Regulation by GPCRs and RTKs |
TRPC1/4/6, TRPV1 | Control of vascular permeability |
TRPC4, TRPV1/4, TRPP1/2, TRPM4 | Regulation of vascular tone |
TRPC4 | Hypoxia-induced vascular remodeling |
TRPC3/4, TRPM2/7 | Oxidative stress response |
TRPC1/4/6, TRPV1, TRPM6/7 | Angiogenesis |
TRPM6/7 | Mg2 + transport (EC proliferation) |
TRPV1–4, TRPM8 | Temperature sensitivity |
Calcium oscillations are mainly due to periodic discharges of intracellular Ca2 + stores. They are triggered by low concentrations of ATP, acetylcholine, bradykinin, histamine, and substance-P. Biphasic increase in intracellular calcium concentration is primed by higher concentrations of these chemical stimuli. Calcium oscillations are frequently accompanied by activation ofKCa1 channels (BK) that provoke oscillatory changes in membrane potential. They require non-selective cation channels, such as TRP channels,39Na + –Ca2 + exchangers, andSERCA pumps.
Vasoactive agents generate Ca2 + waves that travel through the cell at a speed of 5 to 60 μm/s according to stimulus concentration and cellular region [895]. These waves spread through gap junctions.
Members of the transient receptor potential (TRP) family,40 may be involved. Short canonical TRP channels (STRPC) include: (1) receptor-activated cation channels (RACC) that are commonly activated by phospholipase-C; (2) non-selective cation channels (NSC);41 and (3) Ca2 + -selectivestore-operatedchannels.42 Channels TRPC1 and TRPC6 are highly expressed in endothelial cells, but TRPC3, TRPC4, and TRPC7 are weakly produced. Channels TRPC1, TRPC4, and TRPC5 are store-operated channels, whereas receptor-operated channels TRPC3 and TRPC6 are activated independently of store depletion. Multiple heterologous combinations of TRPCs generate tetrameric channels (e.g., TRPC1–TRPC4, TRPC1–TRPC5, and TRPC3–TRPC6 complexes). In bovine aortic endothelial cells,glucose enhances synthesis of canonical transient receptor potential TRPC1 channel, but not that of TRPC3, TRPC4, and TRPC6 [897].
Endothelial Ca2 + -permeable receptor-activated cation channels, activated by an increase in ![$$[\mathrm{{Ca}}^{2+}]_{\mathrm{i}}$$](/wp-content/uploads/2016/09/A216847_1_En_9_Chapter_IEq13.gif) , provide a positivefeedback on their own activation, whereas negative feedbacks are provided by mechanosensitive Ca2 + -permeable and/or cyclic nucleotide-gated channels via NO and non-selective cation channels (Fig. 9.3). Ligand-gated Ca2 + channels, such ascyclic nucleotide-gated channels (CNGC),43P2X receptors,44 and probably Na + –Ca2 + exchangers, provide alternative routes for Ca2 + entry in endothelial cell. Voltage-gated Ca2 + channel is a potential candidate for a sustained Ca2 + influx. Stretch-sensitive Ca2 + channel is inhibited by PKG kinase. Calcium pumps PMCA and SERCA are responsible for efflux and intracellular sequestration.
, provide a positivefeedback on their own activation, whereas negative feedbacks are provided by mechanosensitive Ca2 + -permeable and/or cyclic nucleotide-gated channels via NO and non-selective cation channels (Fig. 9.3). Ligand-gated Ca2 + channels, such ascyclic nucleotide-gated channels (CNGC),43P2X receptors,44 and probably Na + –Ca2 + exchangers, provide alternative routes for Ca2 + entry in endothelial cell. Voltage-gated Ca2 + channel is a potential candidate for a sustained Ca2 + influx. Stretch-sensitive Ca2 + channel is inhibited by PKG kinase. Calcium pumps PMCA and SERCA are responsible for efflux and intracellular sequestration.
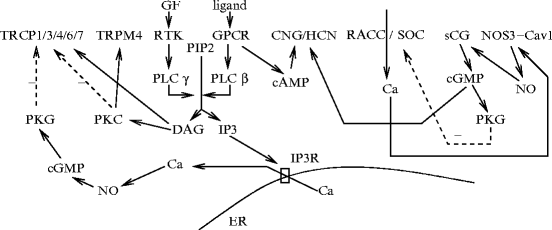
, provide a positivefeedback on their own activation, whereas negative feedbacks are provided by mechanosensitive Ca2 + -permeable and/or cyclic nucleotide-gated channels via NO and non-selective cation channels (Fig. 9.3). Ligand-gated Ca2 + channels, such ascyclic nucleotide-gated channels (CNGC),43P2X receptors,44 and probably Na + –Ca2 + exchangers, provide alternative routes for Ca2 + entry in endothelial cell. Voltage-gated Ca2 + channel is a potential candidate for a sustained Ca2 + influx. Stretch-sensitive Ca2 + channel is inhibited by PKG kinase. Calcium pumps PMCA and SERCA are responsible for efflux and intracellular sequestration.Fig. 9.3
Negative feedbacks resulting from: (1) membrane depolarization on Ca2 + entry caused by cyclic nucleotide-gated (CNG) and/or hyperpolarization-activated CNG K + (HCN) channels and (2) receptor-induced activation of endothelial TRP channels (Sources: [895, 896]). Calcium entry via receptor-activated cation channels (RACC) and/or store-operated Ca2 + (SOC) channels stimulates endothelial nitric oxide synthase (NOS3). Under resting conditions, NOS3 is inactivated due to its binding to caveolin-1. Nitric oxide activates soluble guanylate cyclase (sGC) that produces cGMP messenger. Liganded G-protein-coupled receptors (GPCR) generate cAMP via Gs and adenylate cyclase. Messengers cGMP and cAMP activate CNG and/or HCN channels that cause membrane depolarization, which exerts a negative feedback on RACC and SOC channels. In addition, PKG inhibits RACC and SOC channels. Endothelial TRP channels can be activated by different stimuli to regulate vascular permeability, modulate vascular tone, and cell proliferation and apoptosis. Ligands bind to GPCRs (ATP, bradykinin, acetylcholine, etc.) or receptor protein Tyr kinases (RTK; growth factors) to activate PLCβ or PLCγ that produce DAG and IP3 agents. Messenger IP3 binds to IP3R on the membrane of the endoplasmic reticulum to release Ca2 + from its store. Messenger DAG activates TRPC3, TRPC6, and TRPC7, as well as PKC. PKC inhibits TRPC3, TRPC4, and TRPC6, but stimulates TRPC1 and TRPM4. The NO–cGMP pathway primed by augmented cytosolic calcium level stimulates PKG that inactivates TRPC3, TRPC6, and TRPC7, yielding a negative feedback.
9.5.4.2 Calcium-Gated Potassium Channels
Certain autacoids that are released in response to chemical or mechanical stimuli prime theendothelium-derived hyperpolarizing mechanism that contributes to endothelial control of vascular caliber, particularly in small resistive arteries. This process not only raises the vascular lumen, but also spread the electrochemical signal to coordinate cell behavior along the vessel length (Sect. 9.10).
Potassium influx can hyperpolarize the membrane potential in endothelial cells or smooth myocytes that are coupled via myoendothelial gap junctions. Electrochemical signals that originate from a cell type spread in the blood vessel wall in all directions viagap junctions.Calcium-activated, large-conductance K + (BK) channels hyperpolarize vascular smooth myocytes, thus decreasing vascular tone.Calcium-activated small- (SK) andintermediate-conductance (IK) K + channels control NO synthesis in vascular endothelial cells. Furthermore, they cause depolymerization of cortical actin cytoskeleton. On the other hand, activatedepithelial sodium channels (ENaC) depolarize the membrane potential and provoke polymerization of Gactin monomers in the cell cortex.
Endothelium-derived hyperpolarization mediates vasodilation in the skeletal microcirculation generated byacetylcholine. Acetylcholine can then test the respective contribution of endothelial Ca2 + -dependent channels KCa3.1 (IK) and KCa2.3 (SK) in smooth myocyte relaxation by endothelium-derived hyperpolarization in arterioles [898]. Channel IK is mainly involved in endothelium-derived hyperpolarization, as blockade of SK channel does not have significant effect, although it participates in vasodilation in the absence of IK channel. Moreover, KCa3.1 can not only initiate a response, but also contribute to signal propagation, although it is not mandatory.
9.5.4.3 Voltage-Gated Ion Channels
Endothelial cells produce voltage-gated ion channels [895].Voltage-gated Na + channels are found in endothelia of arteries, veins, and microvessels.Voltage-gated Ca2 + channels localize to capillary endothelium. They belong to CaV1 and CaV3 channel family, as well as voltage-dependent, tiny-conductance, long-opening, SB (sensitive to calcium channel agonist BAY K8644) Ca2 + channels.45 Channel CaV2.3 is activated by a long-lasting depolarization. Hyperpolarization-activated, non-selective cation (K + and Na + ) channels are modulated by nitric oxide.Voltage-dependent, transient outward A-type K + (KV4.2) and delayed rectifier K + channels have been observed.
9.5.4.4 Mechanosensitive Ion Channels
Mechanogated ion channels (MSIC) are required in mechanotransduction, as endothelial cells sense both shear and stretch. Mechanosensitive ion channels can be characterized by their closed-state conformation and gating transition. Ion channel status can be dictated by a coupling withintegrins, cortical cytoskeleton, and G proteins. Opening of shear- or stretch-activated ion channels leads to Ca2 + influx and stimulation of Ca2 + -dependent signaling pathways. Stretch-activated non-selective cation channels of the endocardium and microvascular endothelia increase the intracellular Ca2 + concentration that activates Ca2 + -dependent BK channels, hencehyperpolarizing the endothelial membrane [895].
Mechanosensitive inwardly rectifying K + (KIR),Ca2 + -activated K + 46 (KCa), and probably voltage-dependent K + (KV) channels participate in the setting of the membrane potential, withvolume-regulated anion (VRAC),Ca2 + -activated Cl − (CaCl),cystic fibrosis transmembrane conductance regulator channels (CFTR), andCa2 + -impermeable non-selective cation (NSC) channels (Tables 9.12 and 9.13).
Table 9.12
Potassium channels of endothelial cells (Source: [895]). K + channels induce hyperpolarization, in particular Ca2 + -sensitive K + channels after Ca2 + entry in endothelial cells.
Ion carriers | Features |
|---|---|
Inwardly rectifying | Expression in the macrocirculation; |
K + channels | Location on wetted side of endocardium; |
(KIR2.1) | K + sensor |
(potential threshold based on | |
extracellular K + level) | |
Downregulation by reduction in | |
intracellular ATP and hypoxia | |
Inhibition by phosphatase PP2a, angiotensin-2, | |
vasopressin, vasoactive intestinal polypeptide, | |
endothelin-1, and histamine | |
Hemodynamic stress gating | |
Ca2 + -dependent | EC type-dependent expression; |
K + channels | Ca2 + – and voltage-gating; |
Fluid flow sensitivity (SK and BK) | |
BK | Activation by ryanodine receptor |
IK | Activation by IP3-induced Ca2 + release |
(ATP, acetylcholine, bradykinin) | |
SK | Voltage-independent, calmodulin-dependent |
ATP-sensitive | Activation by low ATP cellular level; |
K + channels | Shear stress-mediated vasodilation (?) |
Stretch-activated | Carriers for K + and Na + |
K + -permeable | |
channels |
Table 9.13
Chloride and sodium channels of endothelial cells (Sources: [895, 896]; CFTR: cystic fibrosis transmembrane conductance regulator channel). Chloride channels prevent hyperpolarization. Volume-regulated anion channels (VRAC) are associated with RhoA activation that restricts the endothelial barrier function. They favors endothelial cell growth and angiogenesis.
Ion carriers | Features |
|---|---|
Chloride channels | |
Ca2 + -activated | EC type-dependent effect on |
Cl − channels (CaCC) | membrane potential |
High-conductance | Activation by β-adrenergic stimuli |
Cl − channels | |
Volume-regulated | Activation by shear and stretch; |
anion channels (VRAC) | participation of annexin, caveolin-1, and RhoA; |
mechanosensor; | |
role in endothelium growth and angiogenesis; | |
decrease intracellular pH | |
CFTR | Reduction in VRAC and CaCC activity |
Shear stress-activated | Sustained membrane depolarization |
Cl − channels | |
Sodium channels | |
Amiloride-sensitive | Microvascular endothelium |
NaV1 | Shear stress sensitivity; |
ERK1/2 activation | |
Na + –K + ATPase | Resting potential |
Na + –Ca2 + exchanger | Ca2 + flux |
Potassium channels participate in endothelium-mediated regulation of the vasomotor tone in adjoining smooth myocytes (Fig. 9.4). ATP-sensitive K + channels may be involved inmechanotransduction.
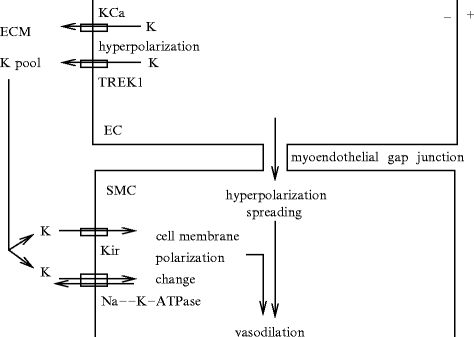
Fig. 9.4
Potassium channels in vascular endothelial cells and smooth myocytes cooperate to regulate the vasomotor tone either via the extracellular matrix (ECM) or myoendothelial junctions.
Volume-regulated anion channel, mainly permeable to Cl − , is activated by shear stress. The complex made of annexin-2 and a regulatory light chain,S100 calcium-binding protein S100a10,47 an element of the cortical cytoskeleton involved in the formation of caveolae, is required for endothelial VRAC activation.
Voltage-activated Na + channels are stimulated by PKC kinase.Epithelial Na + channels may be connected to water transport via aquaporin-4.
Calcium-dependent Cl − channels, outward rectifiers, require ATP agent. Endothelial cell expresses other Cl − channels, includinghigh-conductance Cl − channels, which are quasi-silent in intact cells, andcAMP-stimulated CFTRs.
9.5.5 Hormones and Vasoactive Substances
Functions of the endothelium are affected by hemodynamics. The intracellular calcium concentration in cultured endothelial cells strongly depends on flow pattern.48 Although oscillatory flow does not change [Ca2 + ]i, steady and pulsatile flows increase [Ca2 + ]i, with response dynamics that depend on the flow pattern [899]. The release of the substances active on vasomotor tone depends on shear stress applied on the endothelium (Sect. 9.10). Shear stress might also regulate the secretion of molecules involved in coagulation and fibrinolysis or inhibiting thrombosis (Sect. 9.8). High wall shear stress hinders leukocyte adhesion on the endothelium (Sect. 9.7). The endothelium adapts the vessel wall to the local flow pattern (Sect. 7.11).
A set of signaling messengers and mediators that involves 2 secreted molecules, PDGFbb and TGFβ1, and 4 other proteins, lamin-A, lysyl oxidase, ERK1, and ERK2, contributes to the response of cultured vascular endothelial cells according to the magnitude of applied mechanical stress [900]. Factor PDGFbb that acts on both vascular endothelial cells and smooth myocytes participates in the paracrine control of smooth myocytes by endothelial cells; TGFβ that acts on endothelial cells, but not on smooth myocytes, is implicated in the feedback from smooth myocytes to endothelial cells.
9.5.5.1 Acetylcholine
Acetylcholine is synthesized and stored by choline acetyltransferase in endothelial cells of small brain vessels [901] and rat coronary arteries [904]. Acetylcholine is an NOS3 activator.
In coronary arteries, acetylcholine effect varies according to its concentration and the context (apparently normal or observed atheroma). In normal coronary arteries,vasodilation mediated by NO released from endothelial cells subjected to low acetylcholine concentrations via endothelial muscarinic M3 receptors,49 whereasvasoconstriction mediated by a direct action on smooth myocytes via their muscarinic M3 receptors appears at higher concentrations. At relatively high doses, its direct constrictor effect on smooth myocytes dominates over its vasodilator effect on endothelial cells.
In fact, acetylcholine initiates 2 competing responses in mouse arteries: (1) endothelium-dependent vasodilation mediated predominantly bynitric oxide and (2) endothelium-dependent vasoconstriction mediated bythromboxane-A2 [902].
In atheromatous coronary arteries, vasoconstriction and -dilation occur at low concentration and vasoconstriction happens at high concentrations. In 53% of patients with normal coronary arteriograms, chest pain, and risk factors for coronary artery disease, infusion of acetylcholine causes both constriction and dilation of proximal and distal segments of epicardial coronary arteries [903]. Vasoconstriction and -dilation coexist in different coronary arteries as well as in different segments of a given artery (acetylcholine concentration range 10 − 7–10 − 6 mol/l). In any case, change in epicardial coronary artery caliber in response to acetylcholine does not correlate with change in intramyocardial coronary blood flow.
9.5.5.2 Serotonin and Substance-P
Serotonin andsubstance-P are observed in endothelial cells [905].50 Atheromatous arteries that constrict in response to acetylcholine may dilate upon exposure of substance-P, an endothelium-dependent vasodilator, although dysfunction of the endothelium action is not restricted to muscarinic receptors, but also to other receptors such as tachykinin (substance-P) receptors [903]. In addition, the endothelial response to chemical stimuli is not related to the degree of coronary atherosclerosis.
9.5.5.3 Adrenomedullin
Adrenomedullin is widespread, but at higher concentrations in endothelial cells and vascular smooth myocytes. Adrenomedullin circulates in the plasma at picomolar levels. Elevated plasma concentration of adrenomedullin occur during pregnancy. Strongly vascularized tissues, such as the heart, lungs, and kidneys, release adrenomedullin during inflammation, hypoxia, sepsis, and cardiovascular diseases (myocardial infarction, heart failure, and atherosclerosis), particularly during hypertension.
Adrenomedullin synthesis is regulated via estrogen-responsive elements Adrenomedullin type and concentration vary according to the genetic ground, especially gender, because the adrenomedullin gene is regulated by estrogen. The genetic ground then hinders or favors development of cardiac and renal damages, but in the latter case does not necessarily change the blood pressure [906].
9.5.5.4 Steroid Hormones
Estrogens favornitric oxide production in the endothelium to protect against inflammation, as nitric oxide can induce cysteine S-nitrosylation51 of anti-inflammatory proteins. 17β-Estradiol (E2β), the major estrogen form in the body, causes protein S-nitrosylation in endothelial cells viaestrogen receptor ERα (nuclear receptor NR3a1) and endothelial nitric oxide synthase (NOS3) [907].
In endothelial cells, both basal NO production and 17β-estradiol activation of the PI3K–PKB–NOS3 pathway (estrogen-mediated vascular protection) requireSrc kinase [908]. In the plasma membrane, Src kinase is associated with a truncated, relocalized estrogen receptor-α and NOS3 enzyme. Endogenous 17β-estradiol activates its cognate ERα that stimulates Src kinase. In turn, Src kinase phosphorylates ERα for subsequent plasmalemmal recruitment of Src kinase and ERα and assembling withNOS3, followed by NOS3 activation.
Somesteroid hormones modify endothelial cell behavior. Endothelial cells are insensitive to progesterone and testosterone, but they are softened byestrogens that activate plasmalemmalsodium–proton antiporter.Aldosterone elevates endothelial cell volume and causes growth and stiffness by activating epithelial sodium channelsENaCs, whereas glucocorticoid hormones do not have such effects. In the presence of aldosterone, when plasmasodium level is in the high physiological range, small physiological changes in extracellular sodium concentration stiffens endothelial cells and reduces the release ofnitric oxide. On the other hand, acute increase in extracellular potassium softens vascular endothelium and augments nitric oxide release [909]. Change in cell rheology mediated by potassium and sodium ions mainly involves cortical actin cytoskeleton that switches from gelation to solation according to ambient sodium and potassium concentrations. Potassium causes cortical actin depolymerization, i.e., cortical filamentous actin is transformed into monomeric globular actin that colocalizes with endothelial NOS (NOS3) and heightens its activity.
9.5.5.5 Gasotransmitters
Endogenous gaseous diffusible messengers, or gasotransmitters, include carbon monoxide (CO), nitric oxide (NO), and hydrogen sulfide (H2S), in addition to several reactive oxygen species, that act as intra- and intercellular regulators.
In addition,feedbacks exist among different gasotransmitters that enable the regulation of the action of a given gas by others gaseous mediators [910].
Carbon Monoxide
Carbon monoxide (CO) is a stable, lipid-soluble gas, produced byheme oxygenase (HO) withiron andbiliverdin, using NADPH from heme degradation [911] (Vol. 4–Chap. 10. Other Major Signaling Mediators). Although CO can bind to heme-containing proteins for transient storage and later release, the predominant source is de novo synthesis. It then diffuses according to the local partial pressure gradient. Carbon monoxide binds to the ferrous iron of heme and, then, influences the activity of all heme proteins.
Carbon monoxide not only causes vasodilation, but also supports action of vasodilators and attenuates that of vasoconstrictors such as platelet-activating factor. Endothelial nitric oxide andprostacyclin (PGi2) cooperate with the HO–CO axis to regulate the blood circulation. On the other hand, H2S can inhibit heme oxygenase-2 in endothelial cells [911].
Nitric Oxide
Nitric oxide (NO) a highly diffusible gas that runs very rapidly from its site of synthesis. However, NO is a labile vasodilator with a limited half-life of NO (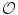 [1 s]); its effect is thus localized close to the site of release. This free radical is highly reactive with other chemical species, notably oxygen, superoxide, and iron-containing hemes that act as NO scavengers. Nitric oxide is able to serve as an auto-, para-, and endocrine signal (Vol. 4 – Chap. 10. Other Major Signaling Mediators).
[1 s]); its effect is thus localized close to the site of release. This free radical is highly reactive with other chemical species, notably oxygen, superoxide, and iron-containing hemes that act as NO scavengers. Nitric oxide is able to serve as an auto-, para-, and endocrine signal (Vol. 4 – Chap. 10. Other Major Signaling Mediators).
[1 s]); its effect is thus localized close to the site of release. This free radical is highly reactive with other chemical species, notably oxygen, superoxide, and iron-containing hemes that act as NO scavengers. Nitric oxide is able to serve as an auto-, para-, and endocrine signal (Vol. 4 – Chap. 10. Other Major Signaling Mediators).Nitric oxide can complex with all transition metals, the so-called metal nitrosyls. Nitric oxide interacts with metalloproteins, especially those that contain hemes, and with thiols. Nitric oxide can act in the form of free radical (NO or NO ⋆ ) or as chemical species, such as nitrosyl (NO + ) or nitroxyl (NO − ) ions or Snitrosothiols (SNO). Hemes either transform NO into nitrate or promote its transformation into Snitrosothiols.
Additional sources of nitric oxide than nitric oxide synthases arise from the cycling of nitrate, nitrite, and nitric oxide. Several pathways modulate the NO –NO
–NO –NO cycling, such as oxidation or reduction by hemoglobin, myoglobin, neuroglobin, xanthine oxidoreductase (XOR), nitric oxide synthase, carbonic anhydrase, cytochrome-C oxidase, cyclooxygenase, microsomal cytochrome-P450, mitochondrial aldehyde oxidase, and cytochrome-C, as well as bacteria, in different tissues under different conditions [912, 913]. Hemoglobin operates at normal pH and oxygen saturation in the vasculature.
–NO cycling, such as oxidation or reduction by hemoglobin, myoglobin, neuroglobin, xanthine oxidoreductase (XOR), nitric oxide synthase, carbonic anhydrase, cytochrome-C oxidase, cyclooxygenase, microsomal cytochrome-P450, mitochondrial aldehyde oxidase, and cytochrome-C, as well as bacteria, in different tissues under different conditions [912, 913]. Hemoglobin operates at normal pH and oxygen saturation in the vasculature.
–NO–NO cycling, such as oxidation or reduction by hemoglobin, myoglobin, neuroglobin, xanthine oxidoreductase (XOR), nitric oxide synthase, carbonic anhydrase, cytochrome-C oxidase, cyclooxygenase, microsomal cytochrome-P450, mitochondrial aldehyde oxidase, and cytochrome-C, as well as bacteria, in different tissues under different conditions [912, 913]. Hemoglobin operates at normal pH and oxygen saturation in the vasculature.Hydrogen Sulfide
Hydrogen sulfide (H2S) is a smooth myocyte relaxant. Whereas cystathionine γ-lyase is the major H2S-producing enzyme in smooth myocytes, 3-mercaptopyruvate sulfurtransferase (3MST) and cysteine aminotransferase (CAT) synthesize H2S from cysteine and α-ketoglutarate in vascular endothelial cells [914].
9.5.6 Adhesion Molecules
Platelet–endothelial cell adhesion molecule PECAM1,52 an inhibitory receptor, contains an intracellular C-terminus with 2 immunotyrosine-based inhibitory motifs (ITIM) that launch inhibitory signaling upon transhomophilic engagement in intercellular interaction (CD31 + cell–CD31 + cell adhesion). Its extracellular motifs are also required for signaling [915]. Molecule PECAM1 participates in the regulation of angiogenesis and preventsleukocyte–endothelial cell adhesion and activation ofplatelets and T lymphocytes. Once PECAM1 is eliminated after cleavage and secretion, inflammation can occur. A truncated extracellular PECAM1 fragment onT lymphocyte surface cannot preclude inflammation, but a PECAM1-derived peptide is able to stimulate this fragment, thereby restoring the PECAM1 inhibitory pathway, thereby repressing inflammation and suppressingatherosclerosis andaneurysm evolution [916]. Molecule PECAM1 primes phosphorylation ofPTPn11 phosphatase in activated leukocytes.
In given environments, cells may express different forms of PECAM1 that exert different functions. For example, it loses its intercellular junction role when endothelial cells are exposed to inflammatory cytokines [915]. In addition, neutrophils can lose PECAM1 surface expression after extravasation.
Oxidized low-density lipoprotein (oxLDL) is an autoantigen that reduces the existence of extracellular PECAM1 and increases the presence of its soluble form in cultures of CD4 + T lymphocytes [915]. In atherosclerotic plaques and tissues surrounding aneurysmal aortas,macrophages have an attenuated activity with reduced production of intracellular peptidases and release of cytokines and chemokines.
In humans, both CD4 + and CD8 + T lymphocytes lose PECAM1 with aging, whereas thrombosis risk augments. Conversely, thrombosis reduces PECAM1 expression, thereby increasing T-cell proliferation. In humans, numbers of peripheral CD4 + , PECAM1 + and CD8 + , PECAM1 + T lymphocytes decrease markedly in patients with abdominal aortic aneurysms, whereas populations of CD4 + , PECAM1 − and CD8 + , PECAM1 − T lymphocytes increase [915].
9.5.7 Miscellaneous
Endothelial cells can also storeArgvasopressin,angiotensin-2,histamine,atrial natriuretic peptide,calcitonin gene-related peptide,neuropeptide-Y, andvasoactive intestinal peptide [680].
9.5.8 Vessel Wall Homeostasis
9.5.8.1 Apoptosis Regulators
Apoptosis regulator through modulating IAP expression (ARIA) synthesized by vascular endothelial cells induces endothelial apoptosis and hampers angiogenesis via proteasomal degradation of inhibitor of apoptosis IAP1 and IAP2 [917].
X-box binding protein XBP1 that is involved in endoplasmic reticulum stress response is highly expressed at areas of atheroma [918]. Sustained XBP1 activation causes endothelial cell apoptosis and favors atherosclerotic lesion (Vol. 6 – Chap. 8. Vascular Diseases).
Humanin (Hn) is a 24-amino acid peptide synthesized in vascular endothelial cells (of both arteries and veins) that protects against oxidative stress and apoptosis [919].53 Exogenous administration of humanin (0.1 μmol) to endothelial cell cultures reduces oxidized LDL-induced formation of reactive oxygen species and apoptosis [919].54 The binding of exogenous humanin to plasmalemmal receptors involved in cell survival pathways and intracellular humanin that binds to BCL2-associated X protein (BAX) can impede BAX pro-apoptotic effects.
9.5.8.2 Angiomotin
In endothelial cells, angiomotin is a double-pass transmembrane protein with an extracellular angiostatin-binding region and intracellular N- and C-termini.
Angiomotin constitutes a family with angiomotin-like-1 and -2 (AMotL1–AMotL2). Members of the angiomotin family possess an angiomotin coiled-coil homology (ACCH) domain that has a high affinity for the monophosphorylated PI(3)P and PI(4)P phosphatidylinositols. Similar to the Bin/amphiphysin/Rvs (BAR) domain, the ACCH domain binds preferentially to curved membranes and induces vesicle tubulation. The angiostatin-binding domain is not present in AmotL1 and AmotL2. The angiomotin C-terminus residues form a PDZ-binding motif.
Angiomotin localizes to tight junctions and endosomes. It is involved in the maintenance oftight junctions andmigration of endothelial cells, as well as insulin sensitivity in skeletal myocytes [920].
Angiomotin is involved in the maintenance of tight junctions. Angiomotin binds to multiPDZ domain-containing protein MuPP1 and its paralog, protein (PALS1) associated with tight junctions (PATJ). These partners couple AMot to tight junction transmembrane proteins, pleckstrin homology (PH) domain-containing RhoA guanine nucleotide-exchange factor PlekHg5, and a large complex involved in the maintenance of apicobasal polarity. Other members of the complex such as protein associated with Lin7 (PALS1) and Lin7 proteins.
Angiomotin colocalizes withzonula occludens protein ZO1 and to a lesser extent withcadherin-555 atadherens junctions [920].
Angiomotin interacts withGTPase-activating RhoGAP17 protein. In the absence of angiomotin, endothelial cells extend lamellipodia simultaneously in several directions, rather than forming a single major lamellipodium at the leading edge [920].
Angiomotin (AMot) was initially identified as a receptor of angiostatin, an anti-angiogenic protein. Angiomotin is required to ensure action of FGF2, VEGFa, and lysophosphatidic acid [920].
Two splice variants are synthesized, short P80AMot56 and P130AMot,57 both produced in vascular endothelial cells [920]. These variants colocalize with tight junction protein ZO1 in endothelial cells.
Variant P80Amot mediates migration stimulation by FGF2 and its inhibition by angiostatin; P130Amot does not intervene. Higher expression of P80Amot corresponds to a migratory phenotype prevalent during angiogenesis; larger production of P130Amot is observed in mature blood vessels [920]. Variant P80Amot causes translocation of P130Amot from tight junction, thereby repressing its stabilizing effect.
Only P130Amot localize to adherens junction. It binds to scaffold membrane-associated guanylate kinase-related protein with inverted domain organization MAGI1b. Only P130Amot binds toactin [920].
Isoforms P80AMot and P130AMot homo- and heterodimerize [920]. In addition, P80AMot colocalizes with the early endosome markerEEA1 (or ZFYVE2) as well as withthe RhoGEF PlekHg5, withARF6 andRab11 in perinuclear recycling endosomes, and with Rab13 GTPase.
9.5.8.3 EGFL7
Multiple EGF-like domain-containing protein EGFL7, or veStatin, is exclusively secreted by endothelial cells to modulate smooth myocyte migration. It may also be involved in the spatial organization of endothelial cells in the angiogenic sprout. It impedes deposition and organization of elastic fibers [921].58 This inhibitor of vascular elastogenesis interacts withlysyl oxidase.59
9.5.8.4 Endosialin
Angiogenic endosialin60 expressed in endothelium binds to fibronectin61 and collagen-1 and -4 to enhance cell–matrix adhesion and cell migration [922]. Endosialin in cells of tumor stroma and endothelium favors tumor angiogenesis and invasion. Endosialin enhances MMP9 activity. In addition, endosialin regulates pericyte–endothelial cell interactions, as its expression is also associated with pericytes.
9.5.8.5 Transglutaminase
Many proteins of the extracellular matrix are substrates oftransglutaminase-2 (TG2) produced by endothelial cells. Transglutaminase-2 affects both matrix deposition and turnover. It promotescollagen-1 synthesis and resistance of the extracellular matrix to degradation by matrix metallopeptidase MMP1, but it downregulates the expression ofcollagen-3 and -4 [923].
9.5.8.6 Heparan Sulfate Proteoglycans
Different structural types ofheparan sulfate are secreted on the cell surface, such as syndecan and glypican, as well as in the basement membrane (thickness of 20–100 nm), such asperlecan, agrin and collagen-18 [924].62 Several heparan sulfate proteoglycans have heparan sulfate chains only under specific circumstances, the part-time heparan sulfates, such as epican (CD44), β-glycan, and testican.
Heparan sulfate proteoglycans interact with manifold proteins, such as growth factors, cytokines, chemokines, peptidases, lipases, and cell adhesion molecules. The vascular endothelium can modulate inflammation by changing the heparan sulfate structure, a given type of heparan sulfate being able to bind or not to a given ligand. Heparan sulfate, in association with interacting proteins, inhibits SMC proliferation [925].
9.5.8.7 Neuregulins
Neuregulins (Nrg1–Nrg4) are cell–cell signaling proteins. They are ligands ofreceptor Tyr kinases of the HER family. Isotype Nrg163 operates in the nervous system, heart, and breast. Neuregulin-1 possesses different isoforms from alternative splicing of the transcript of the NRG1 gene, such as type-1 neuregulin-1,64 type-2 neuregulin-1, or glial growth factors (GGF1–GGF3),65 and type-3 neuregulin-1, or sensory and motor neuron-derived factor (SMDF), in addition to type-4 to type-6 neuregulin-1.
Neuroprotection is based on the neurovascular unit that includes neurons and associated glial cells, vascular endothelium and support cells, and extracellular matrix. Endothelial cells of the human brain microcirculation synthesize neuregulin-1.
Upon ligand binding, Nrg1 receptors HER2 and HER3 are phosphorylated and trigger the PI3K–PKB, Ras–ERK, and JaK-STAT pathways to ensure cytoprotection, particularly during oxidative stress [926]. Because HER2 and HER3 lack ligand-binding and efficient kinase activity, respectively, they form an active HER2–HER3 heterodimer. Other Nrg1 mediators comprise phospholipase-Cγ and protein kinase-C. Neuregulin-1 enhances survival of endothelial cells and can contribute to neuron survival during stroke. Furthermore, neuregulin-1 promotesangiogenesis.
9.5.8.8 Histone Deacetylases – Epigenetic Control by Hemodynamic Stress
Certain class-1 (i.e., HDAC1–HDAC3) and -2a (i.e., HDAC5 and HDAC7)histone deacetylases are sustainably upregulated and accumulate in the nucleus in cultured endothelial cells subjected to a low-steady-component pulsatile flow (modulation rate 8) [927]. On the other hand, a high-steady-component pulsatile flow (modulation rate 1/3) causes phosphorylation-dependent nuclear export of class-2a HDACs.
Histone deacetylases regulate the activity ofNFE2-related factor NRF2 andKrüppel-like factor KLF2 that target many mechanical stress-responsive genes and cell cycle. Transcription factor NRF2 binds to the anti-oxidant response element (ARE) in promoters of many anti-oxidant genes.
Steady flow causes phosphorylation-dependent nuclear export of HDAC5, hence its dissociation frommyocyte enhancer factor MEF2, as well as expression of anti-inflammatory Krüppel-like factor KLF2 and endothelialnitric oxide synthase (NOS3) via sirtuin-1 [927]. Nitric oxide-mediated nuclear shuttling of HDAC4 and HDAC5 relies on the association of protein phosphatasePP2 with the phosphorylatedcalcium–calmodulin-dependent kinase CamK4P–HDAC complex. Phosphorylation of HDAC enzymes by CamK kinases causes HDAC export from the nucleus and cytosolic sequestration by 14-3-3 proteins.
A low-steady-component pulsatile flow uses the PI3K–PKB pathway. It enables the association of class-1 histone deacetylases with NRF2 and that of HDAC3 and class-2a histone deacetylases with MEF2 [927]. Subsequently, NRF2 and MEF2 are deacetylated, thus being unable to bind to ARE element. Consequently, expression of anti-oxidant gene NAD(P)H quinone oxidoreductase-1 (NQO1) and KLF2 is reduced. Moreover, this type of unsteady flow increases the production of cyclin-A, but decreases that of CKI1a cyclin-dependent kinase inhibitor in endothelial cells, thereby inducing their proliferation. In addition, inhibition of KLF2 expression enables elevatedVCAM1 production.
Because a high-steady-component pulsatile flow causes export of class-2a HDACs from the nucleus, it precludes HDAC5/7–MEF2 association, thereby promoting KLF2 expression. Factor KLF2 prevents VCAM1 synthesis, hence leukocyte adhesion. Therefore, VCAM1 expression depends on the modulation rate of the periodic flow [927].
9.5.9 Growth Factors and Regulators of Growth Factor Signaling
Endothelial cells synthesize a huge number of factors (Tables 9.14) to 9.18). They release numerous paracrine growth factors, also calledangiocrine factors. In fact, angiocrine factors comprise secreted molecules as well as plasmalemmal and membrane-anchored compounds that are cleaved and generate liberated variants.
Table 9.14
Endothelial cells are able to synthesize multiple growth factors (Source: [928]). (Part 1) Growth factors (CTGF: connective tissue growth factor; PlGF: placental growth factor; RBP: retinol binding protein; S1PR: sphingosine 1-phosphate receptor). Protein production is influenced by cell localization, type, maturation level (immature or fully-differentiated cell), activity status (resting or activated cell), context (healthy or disease) in a given animal species. Weibel-Palade bodies are secretory granules that store hormones, growth factors, and cytokines, in addition to enzymes, receptors, and adhesion molecules, and release them in response to adequate stimuli. Sprouty proteins (Spry1–Spry4) produced by endothelial cells antagonize fibroblast (FGF), epidermal (EGF), hepatocyte (HGF), and vascular endothelial (VEGF) growth factors, as it is a negative feedback regulator of multiple receptor Tyr kinases.
Type | Receptors |
|---|---|
BMP1–BMP7, BMP9 | BMPR1/2 |
BMP14 (GDF5) | |
GDF15 | |
CTGF | CTGFR |
CSF1/2/3 | CSF1R/2R/3R; erythropoietin EpoR |
HBEGF, | HER1–HER4 |
neuregulin-1, TGFα | |
ephrin-A1–A5/B1–B2 | EPHa1–EPHa8, EPHb1–EPHb4, EPHb6 |
FGF1/2/4/5/7/8/12/16/18/22 | FGFR1/2/3/5 |
HGF | HGFR |
IGF1/2 | Insulin receptor; IGFR1/2 |
NGF | TNFRSF16 |
Neurotrophin-3/4 | NTRK |
PDGFa–PDGFd | PDGFR |
SCF | SCFR |
TGFβ1–TGFβ3 | ALK1, TβR1 (ALK5), TβR2, |
follistatin, FSL1/2, | Cryptic |
inhibins | |
VEGFa–VEGFd | VEGFR1–VEGFR3 |
VEGFa  | Neuropilin-1/2 (VEGF coreceptors) |
PlGF1/3/4 | |
S1P | S1PR1 |
Table 9.18
Endothelial cells are able to synthesize multiple growth factors (Source: [928]). (Part 5) Cytokines, chemokines, and adipokines (CT1R: cardiotrophin-1 receptor; CNTFR: ciliary neurotrophic factor receptor; LIF: leukemia inhibitory factor). Cardiotrophin-1 (CT1), a member of the IL6 family, is a cytokine capable of inducing cardiomyocyte hypertrophy. Ciliary neurotrophic factor (CNTF) is a cytokine that assists in the differentiation and survival of various cell types in the peripheral and central nervous systems. A secreted complex made of cardiotrophin-like cytokine (CLC) and cytokine-like factor-1 (CLF), like CNTF, acts via CNTF receptor.
Type | Receptors |
|---|---|
Cytokines | |
CT1 | CT1R (GP130–LIFR) |
CNTF, CLC–CLF | CNTFR (LIFR–CNTFR–GP130 |
Ifnα/β/γ | IfnaR1, IfngR1 |
IL1–IL3, IL5–IL8, | IL1RA, IL1R2, IL2Rα–IL4Rα, |
IL10–IL20, IL23–IL25, | IL6Rα–IL8Rα, IL10Rα–IL11Rα, |
IL27–IL29, IL32–IL33, | IL13Rα, IL15Rα, IL17Ra, IL18R, |
IL36 | IL20R, IL22R1, IL27Rα, IL33R |
LIF | LIFR |
Oncostatin-M receptor (OSMR) | |
Prolactin | Prolactin receptor |
TNFSF1–TNFSF7, TNFSF10–TNFSF11, | TNFRSF1a–TNFRSF1b, |
TNFSF13b (20), TNFSF15 | TNFRSF3–TNFRSF5, |
TNFRSF6b/11b/12a/14/16/21/25 (12) | |
Chemokines | |
CCL1–CCL5, CCL7–CCL8, CCL11, | CCR1–CCR9 |
CC15–CCL24, CCL26–CCL28 | |
CXCL1–CXCL7, CXCL9–CXCL13, | CXCR3–CXCR4, CXCR6–CXCR7 |
CXCL16–CXCL17 | |
CX3CL1 | CX3CR1 |
XCL1 | |
Adipokines | |
Leptin receptor | |
RBP4 receptor | |
Resistin receptor | |
Visfatin | Visfatin receptor |
Table 9.16
Endothelial cells are able to synthesize multiple growth factors (Source: [928]). (Part 3) Peptides and other types of hormones and growth factors (Cont.; ApoER: apolipoprotein-E receptor; LRP: low-density lipoprotein receptor-related protein; PTHRP: parathyroid hormone-related protein; RXFP: relaxin family peptide receptor; VLDLR: very-low-density lipoprotein receptor). Midkine receptor is the complex formed by receptor protein Tyr phosphatase PTPRb, low density lipoprotein receptor-related protein LRP1, anaplastic leukemia kinase (ALK), and syndecan. Three pleiotrophin receptors exist (N-syndecan, receptor protein Tyr phosphatase PTPRb, and ALK). Neurturin, a member of the GDNF family, like artemin, binds to a complex formed by GFRα2 and Ret receptor protein Tyr kinase.
Type | Receptors |
|---|---|
Midkine | PTPRb–LRP1–ALK–syndecan |
Pleiotrophin receptor | |
Oxytocin receptor | |
Neurokinin-1 (substance-P) | NK1 |
Neuromedin-U | Neurokinin-A/B receptors |
Neuropeptide-Y | Neuropeptide-Y receptors |
Neurotensin receptors | |
Neurturin | GFRa2 |
Prokineticin-1/2 | Prokineticin receptor (PKR) |
Proopiomelanocortin | |
PTHRP | |
Reelin | Reelin receptors (VLDLR, ApoER2 [LRP8]) |
Relaxin | RXFP1 |
Salusin-α/β (torsin-2A) | |
Somatostatin | SstR |
Urocortin-1–urocortin-3 | Urocortin (CRF) receptor |
Urotensin-2 | Urotensin-2 receptor |
Vasohibin | |
Vasopressin | Vasopressin receptor |
Table 9.17
Endothelial cells are able to synthesize multiple growth factors (Source: [928]). (Part 4) Guidance molecules and morphogens (DLL: Delta-like ligand; EDIL: EGF-like repeats and discoidin-1-like domain-containing protein; EDIL3 is an integrin ligand and angiogenesis mediator; sFRP: secreted Frizzled-related protein).
Type | Receptors |
|---|---|
Guidance molecules | |
Netrin-4 | Ntn4R |
Slit-2/3 | Robo1/4 |
Semaphorin-3a/3b/3e–3g, | Neuropilin-1/2 (semaphorin coreceptors) |
semaphorin-4A/7A | Plexin-A1/12/B1/B3/D1 |
Morphogens and remodelers | |
Sonic Hedgehog receptors | |
Notch-1–Notch-4 | DLL1, Jag1/2 |
EDIL3 | |
Wnt1–Wnt3, Wnt5a–Wnt5b, Wnt6, | Frizzled-1–Frizzled-7 |
Wnt7a/8a, Wnt10a–Wnt10b, | sFRP1–sFRP2 |
Wnt13a–Wnt13c, Wnt13 (Wnt2b), | |
Wnt14 (Wnt9a), Wnt15 | |
Miscellaneous | |
Gliomedin | Glucagon-like peptide-1 receptor |
Spondin-1 receptor | |
Neuritin | Neuritin receptor |
Some angiocrine factors can act on stem and progenitor cells, as endothelial cells achieve an instructive vascular niche. Endothelial cells are indeed involved in self-renewal of hematopoietic stem cells. Specific angiocrine factors produced by endothelial cells in response to different stimuli either support self-renewal and proliferation using the TOR–PKB pathway or, due to MAPK coactivation, lineage-specific differentiation of hematopoietic stem and progenitor cells [929]. In particular, Notch ligands by endothelial cells promote HSC proliferation.
Table 9.15
Endothelial cells are able to synthesize multiple growth factors (Source: [928]). (Part 2) Peptides and other types of hormones and growth factors (C1qTNF: C1q and tumor-necrosis factor (TNF)-related protein; GHR: growth hormone receptor; NPR: natriuretic peptide receptor; RAMP: receptor activity-modifying protein). Angiogenic factor with G patch and FHA domains (AGGF1), or vasculogenesis gene on 5q protein (VG5q), is a potent angiogenic factor that may act as an autocrine growth factor promoting slightly endothelial cell proliferation. Mutations of the AGGF1 gene cause Klippel-Trenaunay syndrome characterized by an enhanced angiogenesis. Angiopoietin-1 and -2 and their receptor protein Tyr kinases TIE1 and TIE2 are involved in angiogenesis, Ang1 as a paracrine agonist of TIE2 and Ang2 as an autocrine TIE2 antagonist. In the absence of VEGF, Ang2 antagonizes Ang1 and causes vessel regression; in the presence of VEGF, Ang2 facilitates vascular sprouting. Whereas Ang1 is produced by perivascular cells and stabilize quiescent endothelium, Ang2 is secreted by endothelial cells during angiogenesis. Artemin, an auto- and paracrine factor, is a member of the GDNF family with neurturin, persephin, and glial cell-derived neurotrophic factor. It binds to a complex formed by GPI-anchored coreceptor GFRα3 and Ret receptor protein Tyr kinase Granins are also called chromogranins or secretogranins.
Type | Receptors |
|---|---|
AGGF1 | AGGF1 receptor (integrin-α5β1) |
Angiopoietin-2 | TIE1–TIE2 |
Artemin | Adrenomedullin receptor (RAMP2/3) |
Bradykinin | Bradykinin BR, bombesin BBR |
Cartducin (C1QTNF3) | Cartducin receptor |
CNP | (BNP/CNP) NPR (GuCy2) |
CGRP, gastrin | Cholecystokinin CCKR |
Cortistatin receptor | |
CRHR | |
Endothelin-1/2/3 | ETR |
Metenkaphalin | Op1, Op4 |
Ghrelin | Ghrelin receptor |
GHR | |
Granin (ScG2 or CGc) | Granin receptor |
Kisspeptin | GPR54 |
Angiocrine factors secreted from endothelium also regulate tumor cell proliferation and motility. Angiocrine factorSlit-2, which is inhibited by endothelial EPHa2 receptor, represses tumor growth.
Endothelial cells from different compartments of the vasculature, such as arteries, veins, microcirculation segments, and lymphatic vessels, differ in their gene expression patterns (different phenotypes). In addition, tumor endothelial cells also differ by expression of specific markers.
9.5.9.1 Connective Tissue Growth Factor
Connective tissue growth factor production in endothelial cells can be induced by bioactive lipids, such aslysophosphatidic acid andsphingosine 1-phosphate, and platelets [930].
9.5.9.2 VEGF and Cadherin-5
Endothelial cells produce vascular endothelial (VE)-cadherin, or Cdh5, that binds to cortical partners, such as β-, δ1-, and γ-catenin. Endothelial-specific cadherin-5 clusters build homotypic adherens junctions that enable endothelium stabilization, as they prevent cell growth, proliferation, and apoptosis.
In sparse cells, VEGF quickly induces VEGFR2 clathrin-dependentendocytosis for a long duration.66 Internalized VEGFR2 then associates with PLCγ and PKC to be phosphorylated and to activate the MAPK pathway. The Cdh5–βCtn complex is involved in the development of the cardiovascular apparatus. It links to VEGFR2 to reduce its activity due to coclusteredPTPRj phosphatase [931].67
Confluent endothelial cells hence respond poorly to VEGF factor. Phosphatase PTPRj colocalizes with cadherin-5 and β-catenin at adherens junctions between endothelial cells. It dephosphorylates VEGFR2 and impedes its normal endocytosis that leads to activation ofextracellular signal-regulated kinases ERK1 and ERK2 via PLCγ and PKC enzymes. The Cdh5–VEGFR2 complex limits cell proliferation by preventing activation of ERK1 and ERK2 kinases.
9.5.10 MicroRNAs
SpecificmicroRNAs are involved in various developmental processes, especially mechanisms that support the formation of blood and its circulation, such as cardio- and angiogenesis and hematopoietic lineage differentiation.Dicer (Vol. 1 – Chap. 5. Protein Synthesis) that is constitutively expressed in endothelial cells is required for appropriate vasculo- and angiogenesis in embryos (Chap. 10) [933].Drosha alters endothelial cell function and angiogenesis to a lesser extent than Dicer, althhough Drosha silencing in endothelial cells impairs capillary sprouting and vascular tube formation, two activities associated with endothelial cells.
Endothelial cells synthesize, at least in certain circumstances, more or less specific microRNAs.68 Among them, let7f, miR27b, and miR130a are pro-angiogenic, whereas miR221 and miR222 preclude endothelial cell migration and proliferation, and thus angiogenesis [933].
MicroRNA-210 is produced during hypoxia in endothelial cells to enhance vascular tube formation as well as VEGF-caused migration of normoxic endothelial cells. A group of hypoxia-regulated microRNAs is elicited by hypoxia-inducible factor.
MicroRNA-130a that is produced at low levels in quiescent endothelial cells is upregulated during angiogenesis. It impedes the functioning of anti-angiogenic homeodomain-containing Gax69 and HoxA5 proteins.
On the other hand, anti-angiogenic miR221 and miR222 repress stem cell factor receptor and endothelial nitric oxide synthase (NOS3). Moreover, miR221 and miR222 control the synthesis of other microRNAs for more or less redundant activity.
MicroRNA cluster miR17-92 (miR17, miR18a, miR19a–miR19b, miR20a, and miR92a) is significantly upregulated in tumors to promote angiogenesis in a paracrine manner [933]. These miRs target anti-angiogenic proteins. MicroRNA-18 preferentially suppresses the expression of connective tissue growth factor. MicroRNA-19 impedes thrombospondin Tsp1 activity. The miR17-92 cluster is also involved in the regulation of B-cell differentiation.
MicroRNA-378 improves not only tumor cell growth, but also tumor angiogenesis, as it inhibits SuFu and Fus1 tumor suppressors.
Dicer depletion reduces the expression of microRNAs that control the production ofhigh-mobility group (HMG)-box protein HMGB1 transcriptional suppressor, which targets NOx organizer NOxO2 of the NADPH oxidase complex. MicroRNAs, such as miR21, miR126, and miR155, contribute to vascular inflammation [933]. Overexpression of MiR21 causes neointima formation. MicroRNA-126 inhibits TNFα-stimulated VCAM1 that favors leukocyte adherence to the vascular endothelium. MicroRNA-155 represses the expression of angiotensin-2 AT1R receptor. MicroRNA-155 expression is induced in macrophages by TNFα and Ifnβ for proliferation of granulocytes and monocytes during inflammation.70
In normal adult swine, expression of endothelial microRNA-10a is lower in the inner aortic arch and aorto-renal branches than elsewhere; subsequently those of HoxA1, a miR10a target, and inflammatory biomarkers CCL2 chemokine, interleukin-6 and -8, VCAM-1, and E-selectin rises in the same regions [934]. Transcripts that encode 2 regulators of IκBα degradation, MAP3K7 and β-transducin repeat-containing ubiquitin ligase, contain a miR10a-binding site in their 3′UTR region. MicroRNA-10a thus prevents activation of NFκB and thus expression of a pro-inflammatory endothelial phenotype in the arterial bed.
9.5.11 Angiogenesis Guidance Molecules
New blood vessels arise from existing ones by emergence of tips with non-proliferating tip cells that have a guidance role and are followed by sprouting with proliferating stalk cells. Signaling involved in vessel (and axon) guidance includesDelta-like ligand (DLL)–Notch,71EPH–ephrin,72netrin–Unc5,73 andSlit–Roundabout pathways,74 in addition to VEGF, Sprouty, and Hedgehog (Sect. 10.2; Table 9.19).
Table 9.19
Angiogenesis guidance pathways.
Pathway | Ligand and receptor types |
|---|---|
EPH–ephrin | Ephrin ligands (transmembrane proteins) |
Erythropoietin-producing hepatocyte receptor kinase | |
Netrin–DCC/Unc5 | Secreted netrin-1/3/4/5 |
Deleted in colorectal carcinoma receptor | |
Unc5 receptor homologs | |
Netrin–NtnGR | GPI-anchored netrin-G1/G2 |
Netrin-G single-pass transmembrane receptor | |
Notch–DLL | Notch receptor (single-pass transmembrane heterodimer) |
Notch–Jagged | Delta-like ligands (membrane-bound and soluble proteins) |
Jagged (membrane-bound and soluble proteins) | |
Slit–Roundabout | single-pass transmembrane Robo1–Robo4 molecules |
Secreted Slit ligands |
Robo activated by Slit2 is expressed in stalk cells, but not in tip cells [892]. Receptor Robo1 is expressed by endothelial cells [936]. According to [892], Robo4 suppresses the activity of vascular endothelial growth factor required in angiogenesis (as it induces endothelial cell proliferation and migration, as well as blood vessel wall permeability), hence stabilizing the endothelium. However, Robo4 can have a guidance role in developing vessels [935].
9.5.12 Blood Group Antigens
α(1,2)-Fucosyltransferase Fut1 adds the terminal fucose to form blood group antigen glycan Lewis-Y (CD174) and its precursor glycan H2 (CD173) that are predominantly expressed on erythrocytes and subsets of vascular endothelial cells. Bone marrow and microvascular endothelial cells (but not macrovascular endothelial cells, such as aortic endothelial cells) stimulated by tumor-necrosis factor-α increase their production of blood group glycans-H2 and Lewis-Y. Fucosylated blood group glycans-H2 and Lewis-Y promote tumor angiogenesis [937]. Factors of malignant cells such as TNFα elicit endothelial cell migration to form new blood vessels. Glycans-H2 and Lewis-Y are concentrated on pseudopodial extensions of endothelial cells.
9.6 Transendothelial Mass Transfer
Vascular endothelium is a selective permeable barrier that regulates the transfer of molecules and cells from blood to underlying tissues. Transport across the endothelium depends on relative intramural pressure and concentration gradients. Molecule transport across endothelium is influenced by molecule characteristics: molecular size, charge, shape, and carbohydrate content. In fact, transport properties of the endothelial barrier are sensitive to both its chemical and mechanical environment.
Transport across the endothelium use intra- and paracellular routes. Intracellular route corresponds to vesicular transcytosis. Paracellular route is enabled by breaks in tight (size ∼ 20 nm) and adherens junctions as well as interendothelial leaky cleft uncovered by the glycocalyx (size 80–1330 nm at cell division sites and 15–1000 nm in dying or dead cell zones [938]). Intercellular cleft can be strongly sealed by tight junctions. Mechanical cohesion of the endothelial monolayer is supported by adherens junctions. Adherens junction connects the actin cytoskeleton to the plasma membrane.
Endothelial permeability varies according to the state of the cytoskeleton, i.e., to the width of intercellular spaces that narrows or enlarges with endothelial cell relaxation or contraction.
Flowing blood shears the wetted (luminal) surface of endothelial cells endowed with a glycocalyx that yields the first barrier to transport of solutes and water between blood and tissue, hence modulating the permeability to solutes. Mechanical forces influence transendothelial molecular flux via both intra- and paracellular routes.
9.6.1 Flow-Dependent Transendothelial Transport
Reversible endothelial cell remodeling primed by hemodynamic stresses is associated with an initial increase followed by a gradual decline ( ∼ 40% after 24 h) in electrical impedance of cultured endothelial monolayers. The endothelium permeability significantly increases after relatively short exposure (1 h) to steady flow, but markedly decreases ( ∼ 1/3) after long exposure (7–9 d) [938]. When endothelial cell are subjected to sinusoidal flow with an amplitude ratio smaller than 1, the hydraulic conductivity increases similarly to its response to steady flow. When the modulation rate exceeds 1, i.e., when flow reversal occurs, the hydraulic conductivity does not heighten [938].
Mechanotransduction at the luminal (wetted) endothelial surface provokesnitric oxide synthesis by nitric oxide synthase.Heparan sulfate, the dominantglycosaminoglycan of the endothelialglycocalyx, participates in mechanosensing that mediates NO production in response to flow [939]. Oscillatory flow shear stress (modulation rate 3/2) provokes a greater production of NO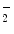 and NO
and NO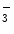 than steady flow.Hyaluronic acid, another glycosaminoglycan of the endothelial glycocalyx, enables mechanotransduction, whereas chondroitin sulfate has no effect on shear-induced NO production and increase in hydraulic conductivity.
than steady flow.Hyaluronic acid, another glycosaminoglycan of the endothelial glycocalyx, enables mechanotransduction, whereas chondroitin sulfate has no effect on shear-induced NO production and increase in hydraulic conductivity.
and NO than steady flow.Hyaluronic acid, another glycosaminoglycan of the endothelial glycocalyx, enables mechanotransduction, whereas chondroitin sulfate has no effect on shear-induced NO production and increase in hydraulic conductivity.In addition, mechanosignaling in response to flow changes in perfused lung microvessels is initiated in caveolae. Flow-preconditioned cells express a 5-fold increase in caveolin and other caveolar proteins at the luminal surface with respect to control (no-flow) condition [944]. Phosphorylation of luminal surface proteins (caveolin-1 and nitric oxide synthase NOS3, which are preferentially localized to caveolae) as well as activation of ERK1 and ERK2 rise upon flow step application in comparison to no-flow condition, more in flow-preconditioned cells than in the absence of flow preconditioning.
Under flow reversing conditions, large NO production limits endothelium permeability via cAMP formation and cAMP-dependent protein kinase (PKA) that controls cytoskeletal tension. On the other hand, the hydraulic conductivity rises under small increases in NO concentration via cGMP production and cGMP-dependent phophodiesterases and protein kinase (PKG) that targets junctional proteins such as tight junctionoccludin.
Mechanotransduction can also exert on the lateral edge of endothelial cells along the intercellular cleft due to convection generated by the pressure difference across the endothelium.
Mechanical stress heightens hydraulic conductivity by raising phosphorylated occludin level and lowering occludin content, hence by tight-junction disassembly [940]. However, exposure of endothelial cells to sustained equibiaxial cyclic strains (1 Hz, 24 h) strengthen tight junction, as it elevates amounts of both occludin and zonula occludens protein ZO1, a linker protein that connects the tight junction to the actin cytoskeleton, as well as reduces occludin phosphorylation, increases ZO1 phosphorylation, and supports occludin–ZO1 association [941]. In addition, maintenance of adherens junction (zonula adherens or belt desmosome) under mechanical stress requires tethering of cadherins to γ-catenin [942]. β-Catenin as well as PECAM1 cannot compensate for the loss of γ-catenin to ensure endothelial barrier integrity under flow. The glycocalyx is implicated in endothelial junction remodeling launched by mechanical stress [943]. Endothelial cells respond rapidly to flow by increasing the amount of cadherin-5 in adherens junctions due to heparan sulfate proteoglycans. In any case, transport through endothelial monolayers subjected to flow depends on endothelial cell type. Nevertheless, the hydraulic conductivity also relies on mechanical stress field in excised arteries.
Most explorations are carried out ex vivo, i.e., in endothelial cells unaccustomed to flow. Experiments on cultured endothelial cells and excised blood vessels do not accurately mimic in vivo conditions.
9.6.2 Transport Mechanisms
Two different transport mechanisms can be defined (Fig. 9.5): (1) an intercellular transport through between-cell junctions for small molecules and (2) an intracellular transport for macromolecules (Vol. 1 – Chap. 9. Intracellular Transport).
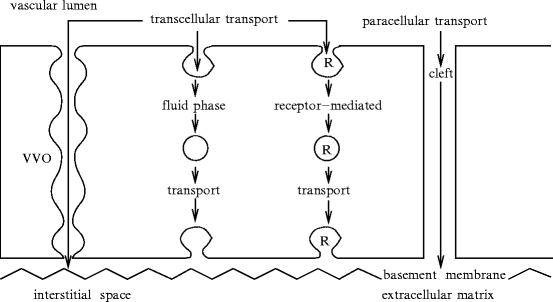
Fig. 9.5
Transport routes in continuous endothelium (e.g., pulmonary, coronary, skeletal muscle, and splanchnic endothelia) comprise para- and transcellular paths (Source: [854]). The paracellular route that uses interendothelial clefts that contain proteins connected to extracellular matrix constituents depends on signaling pathways. Transcytosis is carried out by vesicles that shuttle between the vascular lumen and subendothelial space. Plasma protein size-selective permeability of the endothelial barrier generates a transendothelial protein gradient, the so-called osmotic gradient. Water-insoluble substances are conveyed from blood to interstitium often by specific carrier proteins. Plasma proteins such as albumin serve as chaperones for hydrophobic substances, fatty acids, and hormones. Transvascular flux of solutes and fluid includes transport of small hydrophilic molecules via interendothelial spaces, the permeability of which is regulated, as well as vesicular transit of small hydrophobic solutes through endothelial cells, especially in response to intrinsic and extrinsic stimuli. Small molecules (equivalent radius < 3 nm) such as glucose are transported in interendothelial space, whereas larger plasma proteins such as albumin are conveyed by vesicles filled with receptor-bound (absorptive or receptor-mediated transport) or free solute (fluid-phase transport). Vesiculovacuolar organelles (VVO) formed by fusion of vesicles yield macromolecule transport of growth factors and other regulators. Aquaporins form channels across the lipid bilayer of luminal and abluminal endothelial membranes that are highly selective for water. Inflammatory and angiogenic mediators, such as thrombin, bradykinin, histamine, vascular endothelial growth factor, bind to their receptors and disrupt interendothelial junctions and integrin–matrix protein complexes to form intercellular gaps and open the interendothelial barrier.
Vascular wall permeability varies according to regions. Wall filtration measurements show that arterial and venous beds are much less efficient than microvascular compartments. Microvascular endothelia regulate tissue fluid balance and supply nutrients, as these semipermeable barriers control paracellular and transcellular transports between blood and interstitial space. Vascular endothelia carry solutes, the molecular radius of which range from 0.1 nm (sodium ion) to 11.5 nm (immunoglobulin-M) [854]. Endothelial permeability decreases by four orders of magnitude when molecular radius increases from 0.1 to 3.6 nm. Low-molecular-weight molecules (water, hexoses, such as glucose, mannitol, and fructose, amino acids, and urea) go up endothelial clefts, i.e., the paracellular permeation path of continuous endothelia. When molecular radius rises from 3.6 to 6 nm, endothelial permeability remains nearly constant, as the transport shifts to transcytosis.
9.6.2.1 Intercellular Transport – Normal and Leaky Regions
The junctions between endothelial cells act also as a selective barrier to the egress of water and hydrophilic solutes from blood circulation. Endothelium can be divided into 2 parts according to the state of endothelial cells and their junctions: (1) normal endothelial cells, the most numerous, with normal junctions; and (2) leaky endothelial cells, either mitotic or apoptotic endothelial cells of random location, with leaky junctions. The dynamic regulation of endothelial integrity relies on cell migration coordination and re-establishment of cellular junctions. These processes are regulated by the actin cytoskeleton, and thus, in particular, byRho GTPases and their effectors.
Macromolecular permeability between endothelial cells is regulated by tight junctions, the most apical component of the intercellular cleft, and the thinnest intercellular space, making intimate contact between adjacent endothelial cells. Most water and hydrophilic solutes that are small enough (lower than the albumin dimension) can cross the orifices of tight junctions in the endothelium clefts.
The spaces between several adjoining endothelial cells such astricellular corners, where the borders of three endothelial cells meet, are more permeable regions because endothelial tight junctions are discontinuous at the corners [945]. Discontinuities are observed for occludin, ZO1, cadherin, and β-catenin at the tricellular corners [946]. In addition, 70% of neutrophil transendothelial migration occurs at tricellular corners.
Leaky endothelial cells favor big molecule transport. Endothelium permeability is, indeed, enhanced when endothelial cells have high turnover rates such as in atherosclerosis [947]. Moreover, hypoxemia induces EC apoptosis, and thereby increases macromolecular transport across the vessel wall via leaky junctions. The normal junctions are modeled as circular section spaces around endothelial cells with a pore set in its central part, and leaky junctions as rings around leaky endothelial cells [948].
9.6.2.2 Transcellular Transport
The highest transfer flux between the vascular lumen and extracellular medium occurs by vesicles (size 80 nm). Vesicles can fuse to one another and possibly with cytosol organelles to form tubules. Vesiculovacuolar organelles (VVO; caliber 80–140 nm; length 1–2 μm) that are formed by fusion of vesicles or vacuoles ( ∼ 80–360 vesicles) in continuous endothelia of venules build channels through the cytoplasm. These channels link the luminal edge to abluminal or lateral endothelial cell surface. Vesiculovacuolar organelles convey macromolecules, such as ferritin, vascular endothelial growth factor, serotonin, and histamine, that can bind VVOs by means of receptors [949]. Moreover, vascular endothelial growth factor favors transcellular route via VVOs. Stomatal diaphragms between adjacent, fused vesicles within VVOs can act as barriers between ends of VVOS. Membrane-bound tubules can create transendothelial channels for transfer of large plasma molecules [950]. Vesiculotubular structures specialize according to molecule types.
A significant amount of water (up to 40%) crosses the endothelial barrier through an aquaporin-based transcellular route.
Circulating proteins can interact with the endothelial cell surface to enhance or hinder transcapillary exchange. Endothelial plasmalemmal receptors or transporters exist for plasma molecules, such as albumin, a multifunctional carrier (for fatty acids, sterols, amino acids, hormones, metal ions, etc.), insulin, orosomucoid, an acute-phase glycoproteic α-globulin, ceruloplasmin, a copper-carrying ferroxidase, transferrin, lipoproteins, and glucose, among others.
Many amino acid transporters exist in endothelial cells. Cationic amino acids, such as arginine (Arg), histidine (His), lysine (Lys), and ornithine (Orn),75 are transported through the cellular membrane by 4 transport systems (y + , y + L, b0 + , and B0 + ; Table 9.20). Both y + and y + -like transport systems are inhibited by arginine and lysine.
Table 9.20
Transmembrane transport of cationic amino acids. Amino acid transporters are made of a light and a heavy chain.
SLC alias | Transport system alias |
|---|---|
L transport system | |
SLC7a5 | LAT1 (light chain) |
SLC7a8 | LAT2 |
SLC3a2 | 4F2HC (heavy chain) |
y + transport system | |
SLC7a1 | CAT1 (light chain) |
SLC7a2 | CAT2 |
SLC7a3 | CAT3 |
SLC7a4 | CAT4 |
SLC3a2 | 4F2HC (heavy chain) |
y + L transport system | |
SLC7a7 | y + LAT1 (light chain) |
SLC7a6 | y + LAT2 |
SLC3a2 | 4F2HC (heavy chain) |
b 0+ and B 0+ transport system | |
SLC7a9 | BAT1 (light chain) |
SLC3a1 | rBAT (heavy chain) |
x 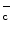 transport system transport system | |
SLC7a11 | xCT (light chain) |
SLC3a2 | 4F2HC (heavy chain) |
asc transport system | |
SLC7a10 | Asc1 (light chain) |
SLC3a2 | 4F2HC (heavy chain) |
The Na + -independent L transport system carries large neutral amino acids with branched or aromatic side chains, such as leucine, isoleucine, valine, phenylalanine, tyrosine, and tryptophan. L-type (large neutral) amino acid transporters include LAT1, or SLC7a5 carrier, LAT2, or SLC7a8 carrier.
The y + transport system is insensitive to neutral amino acids (NAA) and does not depend on pH. It mediates the bidirectional CAA transport. The y + transfer system is based on cationic amino acid (CAA) transporter (CAT) that carries Larginine, a substrate fornitric oxide [951]. The y + transport is mediated by ubiquitous plasmalemmal CATs (CAT1, or SLC7a1, CAT2a–CAT2b, CAT3, and CAT4).
The y + L transport system is sensitive to neutral amino acids. It has a reduced affinity in the absence of Na + cation. It recognizes cationic amino acids in the absence of Na + , but it requires sodium to interact with neutral amino acids. The exchangeable y + -like transport is mediated by y + L amino acid transporters (y + LAT), such as y + LAT1 (SLC7a7) and y + LAT2 (SLC7a6).
The B0, + transport system, which is sensitive to cystine, is another route that use B0, + AT1 carrier. The substrate specificity of B0, + and b0, + transport systems is similar, but the former also accepts alanine and serine. The B0, + transport is mediated by SLC3a1 carrier (cystine, dibasic and neutral amino acid transporters), or B0, + -type amino acid transport76 Activator of cystine, dibasic and neutral amino acid transport possesses variants (SLC3a1 variant-B2–SLC3a1 variant-G2).
The b0, + transport system recognizes cationic and neutral amino acids with a similar affinity. It carries amino acids independently of sodium. The b0, + transport system depends on both Na + and Cl − ions. It conveys cationic and neutral amino acids with the highest affinity for hydrophobic amino acids. The b0, + transport is mediated by SLC7a9 carrier (glycoprotein-associated amino acid transporter light chain, b0, + transport system), a.k.a. cationic amino acid transporter of y + transport system and B0, + -type amino acid transporter BAT1, B0, + AT, and b0, + -type amino acid transporter (b0, + AT).77
Transcripts (mRNAs) of amino acid transporters CAT1, LAT1, LAT2, y + LAT1, y + LAT2, and b0, + AT are expressed together with the heterodimeric glycoproteins SLC3a2 carrier (activator of dibasic and neutral amino acid transport)78 and rBAT transporter.
In fact, the amino acid transporter systems L, y + L, x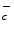 , asc, and b0, + represented by transporter light chains require interaction via a disulfide bridge with the type-2 membrane glycoproteins — heavy chains — for transmembrane transfer. System L, y + L, x
, asc, and b0, + represented by transporter light chains require interaction via a disulfide bridge with the type-2 membrane glycoproteins — heavy chains — for transmembrane transfer. System L, y + L, x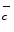 , and asc require SLC3a2, whereas system b0, + requires rBAT subunit.
, and asc require SLC3a2, whereas system b0, + requires rBAT subunit.
, asc, and b0, + represented by transporter light chains require interaction via a disulfide bridge with the type-2 membrane glycoproteins — heavy chains — for transmembrane transfer. System L, y + L, x, and asc require SLC3a2, whereas system b0, + requires rBAT subunit.The x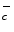 transport system relies on SLC7A11 carrier, also called cystine/glutamate transporter xCT and anionic amino acid transporter light chain.79 The asc transport system is based on neutral amino acid transporter SLC7a10 that corresponds to the light chain of the dimeric carrier.
transport system relies on SLC7A11 carrier, also called cystine/glutamate transporter xCT and anionic amino acid transporter light chain.79 The asc transport system is based on neutral amino acid transporter SLC7a10 that corresponds to the light chain of the dimeric carrier.
transport system relies on SLC7A11 carrier, also called cystine/glutamate transporter xCT and anionic amino acid transporter light chain.79 The asc transport system is based on neutral amino acid transporter SLC7a10 that corresponds to the light chain of the dimeric carrier.In endothelial cells, arginine uptake does not depend on extracellular Na + and Cl − , but is inhibited by neutral amino acid glutamine and leucine. In addition, molecules can be modified during endothelial transit.
Intracellular transport can begin with receptor binding.80 Once bound to their receptors, the molecules assemble in a patch that then bulges to form a vesicle. From the starting membrane granulation to its fusion with target membrane, the vesicle carries the molecules. Such types of shuttles also transport manufactured molecules from theendoplasmic reticulum to theGolgi body, from which are launched vesicles for immediate extracellular release, vesicles for signal-dependent secretion, and digestive lysosomes.
The intracellular receptor-mediated transport is aimed at transfering molecules either to feed endothelial cell (endocytosis) or across the cytoplasm to supply wall tissues with nutrients and growth factors from the blood (transcytosis). Endocytosis allows not only nutrient uptake, but also membrane maintenance. After internalization, main routes lead to degradation, recycling,81 transient sequestration in endosomes, or transcytosis. Pinching off of vesicles requires dynamin. Vesicular traffic is controlled.
Clathrin-mediated endocytosis, which involves actin, captures receptors, ligands and extracellular fluid. Caveolae-mediated endocytosis82 is involved in receptor-mediated endocytosis and the transport of blood macromolecules. Caveosomes transport albumin across the endothelium. Vesicles used for intracellular transport are also involved in signal transduction. Caveolin of the caveolar membrane binds to certain signaling molecules and can be involved in angiogenesis and apoptosis. Actin and microtubules are implicated in raft-mediated endocytosis.
9.6.2.3 Caveolae in Transcytosis
Transcellular transport, or transcytosis, corresponds to endocytosis followed by exocytosis at apical and basal plasma membranes of endothelial cells, respectively.Caveolae correspond to a large fraction of the plasma membrane of endothelial cells (10,000–30,000 per cell; size 50–100 nm) [854]. Caveolae trigger transport of plasma proteins, fatty acids, hormones, and other signaling molecules.
Transcytosis involves a sequential series of events: budding, fission, translocation, docking, and fusion of caveolae to the abluminal membrane. Transcytosis is initiated by interaction of plasma proteins with specific docking molecules in plasmalemmal caveolae that are released upon scission. Caveola-mediated transcellular route uses multiple caveola-associated regulators, such as dynamin and intersectin.
Constituents of endocytic vesicles includecaveolin in addition to dynamin and intersectin. The caveolin family includes 3 gene products (Cav1–Cav3). Caveolin-1 is the major component of caveolae. Caveolin-1 has 2 isoforms. Caveolin-1 and -2 are coexpressed in many cell types, especially in endothelial cells, smooth myocytes, fibroblasts, and adipocytes, whereas caveolin-3 is a muscle-specific type. Endothelial cells from brain microvessels as well as type-2 alveolar epithelial cells express caveolin-1, but have relatively few caveolae. Otherwise, caveola number is correlated with caveolin-1 concentration.
Caveola genesis that requires caveolin-1 occurs by plasmalemmal lipid remodeling into specialized membrane nanodomains rich in cholesterol and sphingolipids. Caveolin-1 binds with high affinity to cholesterol, sphingolipids, sphingomyelin, and glycosphingolipids [854]. Plasmalemmal cholesterol regulates expression of caveolin-1 and caveola assembly. Caveolin-1 forms oligomers of 14 to 16 monomers in caveolae.
Caveolin-1 self-assembly begins in the endoplasmic reticulum. Post-translational modifications of monomeric caveolin-1 (acylation, palmitoylation, and phosphorylation) and conjugation of glycosphingolipids facilitates caveolin-1 oligomerization. Cholesterol is needed for caveolin-1 oligomerization and recruitment to the plasma membrane. Oligomerized caveolin-1 stabilized by membrane cholesterol generates caveolae owing to cofactors such as small Ras GTPase, whereas large GTPase dynamin recruited by intersectin releases caveolae from the plasma membrane.
Caveolin-1 interacts with diverse signaling molecules, such as EGFR and PDGFR receptors, Gq and other GTP-binding proteins, Src and PKCα kinases,IP3R andTRPC1 Ca2 + channels, andnitric oxide synthase NOS3. Caveolin-1 inhibits NOS3 enzyme. Caveolin-1 regulates activity ofRac GTPase andmatrix metallopeptidase.
Caveolin-1 favors association ofβ-catenin withcadherin, thereby promoting the assembly of adherens junction complex and contributing to the endothelial paracellular barrier.
In endothelial cells, cortical actin barrier that is integrated with the plasma membrane must be crossed by vesicles that carry various types of molecules. However, actin participates in caveola-mediated transport, as localized actin polymerization occurs at sites of caveola fission.Dynamin interacts with actin-polymerizing proteins of cortical actin filaments, such as the ARP2–ARP3 complex, cortactin, filamin, profilin, and WASP, as well as adaptor GRB2. Therefore, dynamin is able to control actin dynamics required for transport of released caveolae. Actin-related nanomotor myosin serves to carry vesicles in the cell periphery in both anterograde and retrograde direction with respect to plasma membrane. Microtubules also drive caveola motion within the endothelial cell owing to their nanomotors kinesins and dyneins.
Dynamin causes caveolar fission. Homotetrameric dynamin undergoes GTP-dependent self-assembly that increases its intrinsic GTPase activity. Dynamin oligomers generate a constricting force around vesicle collars that leads to membrane fission and caveola internalization.
Intersectins regulate membrane fission and fusion of endothelial caveolae. Intersectins are dynamin partners and regulators. Intersectin isoforms comprise nerve-specific intersectin-1 and ubiquitous intersectin-2. The latter has 2 splice variants, short (intersectin-2S) and long (intersectin-2L) forms.83 Intersectin-2 is located at necks of caveolae, where it can sequester dynamin dimers or tetramers [854].
Scaffold intersectin binds to Ca2 + , phospholipids, inositol phosphates, WASP,RasGEF Son of sevenless,CDC42 and Rac1 GTPase-activating protein RhoGAP31, soluble Nethylmaleimide-sensitive factor attachment protein receptor (SNARE), synaptosome-associated protein SNAP23 and SNAP25, and synaptojamin. Unlike dynamin, intersectin remains associated with vesicles following fission.
Caveolae requirevSNAREs and tSNAREs on vesicle and target membranes, respectively. Vesicle docking and fusion involvesyntaxin,Rab5, vesicle-associated membrane protein (VAMP),84 and Nethylmaleimide-sensitive factor (NSF). Molecule NSF binds SNARE complex via soluble α- or βSNAP. Hexameric ATPase NSF and αSNAP are SNARE regulators that catalyze SNARE complex disassembly following membrane fusion and liberate SNARE monomers for recycling [854]. Small GTPases of the RAB family regulate cellular transport. Endothelial cells express Rab1 to Rab9, Rab11, Rab13 to Rab15, Rab18, Rab22, and Rab30 [854].
Caveola-associated proteins can regulate transcytosis. Endothelial macromolecular transcytotic complex that comprises caveolin-1, dynamin, Rab5 GTPase, Nethylmaleimide-sensitive factor, and syntaxin is involved in caveola fission, targeting, docking, and fusion with the plasma membrane.
Kinase Src allows formation of the caveolin-1–dynamin complex after phosphorylation of both dynamin and caveolin-1. Balance between tyrosine phosphorylation and dephosphorylation of caveolin-1 and dynamin controls endocytosis.Protein kinases PKCα, PKCβ, and PKCδ lodge in caveolae. Activation of these PKC isoforms as well as PKCε and PKCγ prevents caveola internalization [854].Phosphatidylinositol 3-kinase is also involved in endocytosis.
Caveolae containCa2 + -signaling elements, such as IP3R, store-operated Ca2 + channels, transient receptor potential channels TRPC1 and TRPC3, and Ca2 + ATPase [854]. Increase in intracellular Ca2 + concentration induces exocytosis of Weibel-Palade bodies.
9.6.3 Endothelial Permeability and Vascular Compartments
Structural and functional differences among endothelia that coat successive compartments of the vasculature are associated with changes in transport rate. Endothelium structure is a first agent that can explain differences in transport along the vasculature. Postcapillary venules have intercellular gaps of 3.0 nm, whereas clefts of arterioles and capillaries are impermeable to molecules with size greater than 2.0 nm [854].
Endothelial cell expression and extracellular matrix composition vary according to the vascular territory. Expression of Lin1, Isl1, and Mec3 (LIM) kinase(LIMK),myosin light chain kinase (MLCK), guanine nucleotide-exchange factor Vav for Rho GTPase, and myosin is higher in endothelial cells of microvessels than in larger vessels [854]. Cytoskeletal reorganization and intercellular and cell–matrix attachment remodeling regulate endothelial transport. Moreover, caveola density is the highest in capillaries.
Endothelial cells are coupled to the extracellular matrix that influences transport. Degradation of fibronectin, collagens, or proteoglycans indeed increases ECM permeability. The extracellular matrix contains collagen-4, laminin, fibronectin, entactin, chondroitin sulfate, heparan sulfates (perlecan and syndecan), and matrix-associated proteins, such as thrombospondin and adhesion antagonist secreted protein acidic and rich in cysteine (SPARC). Matrix proteins laminin and collagen-4α1 and -4α2 are associated with microvessel endothelia, whereas a greater contribution of fibronectin and collagen-5α1 and -5α2 is observed in large vessel endothelia [854]. Specific interactions between endothelial cell adhesion molecules and extracellular matrix constituents can modify endothelial permeability from a vascular compartment to the other.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


