Fig. 26.1
Nitric oxide biosynthesis and signal transduction in the vasculature. Nitric oxide (NO) synthesis occurs in endothelial cells following a reaction catalysed by the endothelial NO synthase (eNOS) converting L-arginine to L-citrulline. Tetrahydrobiopterin (BH4) serves as a cofactor in this reaction. Upon its release from the endothelial cells, NO diffuses to neighbouring cells and activates a soluble guanylyl cyclase (sGC) that transforms the guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). Binding of cGMP to its downstream effector protein kinase G (PKG) triggers the phosphorylation of several target proteins that mediate the physiological responses of NO. These responses include the inhibition of growth factor- and vasoactive peptide-induced mitogen-activated protein kinase (MAPK) signalling and phosphatidylinositol-3-phosphate (PI3-K) signalling as well as the inhibition of the Gq-protein-mediated activation of the nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) activity. As a result, PKG-mediated NO activity leads to an attenuation of the proatherosclerotic events such as oxidative stress, cell proliferation and migration, aberrant protein synthesis and gene transcription. In addition, PKG also mediates the vasodilatory responses to NO
NO exerts its biological effects through the activation of a soluble guanylyl cyclase (sGC) by binding to its NO-specific pentacoordinate ferrous heme, causing an allosteric modification that results in increased catalytic activity [11, 12]. GC in turn catalyses the conversion of intracellular guanosine triphosphate (GTP) to cyclic guanosine 3′5′-monophosphate (cGMP) [13]. cGMP acts as a second messenger and transduces NO signal into intracellular events via the activation of the cGMP-dependent protein kinase (PKGs) [13]. Mammalian tissues express two different types of PKGs, type 1 (PKG-1) and type 2 (PKG-2) [14, 15]. PKG-1 is the predominant type found in cardiovascular tissues where it has been shown to mediate the anti-proliferative responses of cGMP [15–18]. PKG-1 modulates the activity of its downstream targets through serine/threonine phosphorylation. These targets include inositol-1,4,5-triphosphate (IP3) receptor, phospholamban, troponin, myosin light chain phosphatase and c-Raf kinase [13, 19–23]. PKG-mediated actions of NO include the decrease in intracellular calcium levels and smooth muscle relaxation [24–26]. However, some actions of NO can be independent of the sGC/cGMP/PKG cascade; these include changes in cyclic adenosine monophosphate (cAMP) signalling [27] or the deleterious production of peroxynitrite radical (ONOO−) [28–30]. In fact, elevated concentrations of NO can react with O2 − that results in the formation of ONOO−, a potent oxidant with the potential to disrupt protein structures by nitrating their tyrosine residues [31] (Fig. 26.2). Therefore, NO can also modulate the activity of several proteins either by nitrosylation of their thiol residues, by nitration of tyrosine or by oxidation [26]. Furthermore, NO has been reported to inactivate NADPH oxidase by antagonising its assembling process, thereby inhibiting ROS generation [32]. However, in physiological settings, NO-induced activation of sGC remains the most important pathway in mediating NO responses [24].
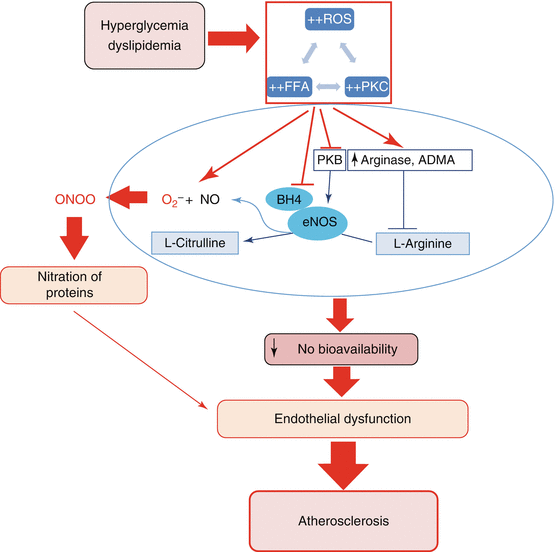
Fig. 26.2
Schematic model depicting the mechanisms leading to a decrease in NO bioavailability and subsequent vascular endothelial dysfunction. Under hyperglycaemic and dyslipidaemic conditions, both increased levels of free fatty acids (FFA) resulting from the metabolism of triglycerides and increased levels of glucose lead to an aberrant activation of the protein kinase C (PKC) that acts as a mediator of NADPH oxidase activation, the latter leading to reactive oxygen species (ROS) generation. ROS in turn can exacerbate protein kinase C (PKC) activation or FFA accumulation. The consequences of this crosstalk include quenching of bioavailable nitric oxide (NO) by superoxide anion to produce deleterious peroxynitrite (ONOO), oxidation and catabolism of the endothelial NO synthase (eNOS) cofactor BH4, inhibition of the protein kinase B (PKB)-mediated activation of eNOS and compromised utilisation of L-arginine due to an increase in arginases and asymmetric dimethylarginine (ADMA). As a result of these effects, there is a decrease in NO bioavailability leading to vascular endothelial dysfunction, the early feature of atherosclerosis
26.3 Action of NO in the Vasculature
Numerous vasculoprotective actions of NO have been reported and are reviewed elsewhere [33]. Briefly, in addition to its vasodilating effect, the anti-atherosclerotic actions of NO include inhibitions of atherogenesis [34], thrombocyte aggregation [35] and VSMC proliferation [36–38] and migration [39]. NO may also antagonise the physiological and pathophysiological effects of growth factors and vasoactive peptides such as epidermal growth factor (EGF) [36], platelet-derived growth factor (PDGF) [40] and basic fibroblast growth factor (bFGF) [39] by interfering with some of the signalling events induced by these factors [34, 37, 39, 41, 42] (Fig. 26.1). Indeed, NO has been reported to inhibit protein synthesis and c-Fos expression in response to endothelin-1 (ET-1) [43–45] and angiotensin II (Ang II) [46]. Early reports showed that NO was able to suppress Ang II-induced activation of three mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinase 1/kinase 2 (ERK1/ERK2), p38MAPK and c-Jun N terminal kinase (JNK) [47] as well as the non-receptor tyrosine kinase Pyk2 [48] in cardiac fibroblasts. In addition, Ang II-induced proliferation of VSMC was found to be attenuated by leptin via NO-mediated signalling [49]. Not surprisingly, due to an eventual reduction of NO bioavailability in hypertension, this effect of leptin was attenuated in VSMC isolated from 10-week-old spontaneously hypertensive rats [49].
26.4 NO Bioavailability and Vascular Endothelial Dysfunction
Multiple lines of evidence support that the processes that lead to either a reduction in NOS substrate L-arginine, modulation of endothelial NOS (eNOS) activity or expression or direct quenching of newly synthesised NO, may account for vascular endothelial dysfunction via their influence on NO bioavailability. It was shown that L-arginine supplementation in monkeys reversed endothelial dysfunction induced by hypercholesterolemia, supporting strongly that a depletion of the NOS substrate could result in altered NO synthesis and contribute to pathologies related to atherosclerosis [50]. It was further revealed that ROS such as hydrogen peroxide and peroxynitrite, as well as oxidised lipoproteins, could induce the expression and the activity of arginase in aortic endothelial cells via the RhoA/Rho kinase pathway amongst other unspecified pathways [51–54]. Consistent with a potential role of arginine deficiency in impaired endothelial function, hyperglycaemia has been reported to enhance arginase activity [55]. By metabolising L-arginine into urea and ornithine, arginase contributes to a decrease in arginine levels resulting in diminished NO biosynthesis and has been implicated in the pathogenesis of vascular dysfunction in T2DM, hypertension and ageing [56–60]. Consequently, arginase inhibitors were shown to improve endothelium-dependent vasorelaxation in patients afflicted with T2DM and coronary artery disease [61] as well as to alleviate hypertension associated with T2DM and metabolic syndrome [62, 63]. Furthermore, in addition to their direct influence on arginine degradation, ROS have also been shown to induce the accumulation of methylated arginine (asymmetric dimethylarginine (ADMA)), endogenous competitive inhibitors of L-arginine binding to the eNOS [64, 65]. ADMA was demonstrated to be upregulated in diabetes mellitus, in association with increased levels of ROS [66, 67]. Indeed, some antioxidants exert their beneficial effects on vascular function via their ability to modulate the level of ADMA [68–70]. However, arginases and ADMA are also able to induce ROS generation through their interaction with eNOS [71, 72]. Therefore, their role as mediators of ROS-dependent impairment in NO bioavailability in endothelial dysfunction remains unclear.
Since BH4 is a key cofactor responsible for the catalytic activation of eNOS activity, impairment in BH4 levels has been suggested to play an important role in regulating the bioavailability of NO [73]. Consistent with this, transgenic over induction of BH4 synthesis has been shown to preserve NO-mediated endothelial function in diabetic mice [74] and to reduce atherosclerosis in apolipoprotein E-knockout (ApoE−/−) mice [75]. Also, BH4 supplementation was shown to reverse endothelial dysfunction in healthy subjects subjected to transient hyperglycaemia [76] as well as in diabetic patients [77], suggesting that BH4 depletion may also account for endothelial dysfunction in T2DM. ROS such as superoxide and peroxynitrite are able to rapidly oxidise BH4 bound or not to eNOS leading to BH4 catabolism and depletion [78]. In addition, oxidation of BH4 results in the formation of dihydrobiopterin (BH2), which binds to eNOS and generates superoxide anion but not NO [79, 80]. Moreover, oxidative stress-mediated eNOS uncoupling can also occur via direct S-glutathionylation of eNOS [81–83]. In addition, ROS can decrease NO bioavailability by directly quenching free NO to form peroxynitrite [84]. Thus, both substrate and cofactor deficiency and excessive ROS levels can modify NO bioavailability and impair endothelial function leading to atherosclerotic vascular disease (Fig. 26.2).
26.5 Hyperglycaemia, Insulin Resistance and NO System
Diabetes is a major risk factor for vascular disease such as atherosclerosis, and hyperglycaemia has been suggested to contribute to vascular endothelial dysfunction-associated vascular diseases. Hyperglycaemia has also been shown to induce the generation of ROS by activation of NADPH oxidase system. ROS, by their ability to activate multiple signalling pathways linked with cell growth and proliferation and by modifying NO bioavailability, can contribute to vascular endothelial dysfunction as outlined in the previous section. In addition, insulin has been reported to increase eNOS gene expression and protein activity resulting in increased NO bioavailability and is known to exert vasodilatory actions [85–88]. Insulin has also been shown to activate eNOS by inducing its phosphorylation on the serine residues, Ser615 and Ser1177 [89]. Consistent with this, it was reported that loss of insulin-mediated vasodilation in VSMC isolated from insulin-resistant diabetic Goto-Kakizaki rats could be reversed by overexpressing a constitutively active form of protein kinase B (PKB) [90]. Thus, diabetic atherosclerosis has been attributed to a defect in PI3-K/PKB/eNOS pathway. In contrast, a group reported that constitutively active PKB failed to stimulate the catalytic activity of eNOS immunoprecipitated from human coronary endothelial cells incubated in high glucose and high glucosamine [91]. It was suggested from these studies that in high glucose conditions, some other mechanisms may directly affect eNOS activity in a PKB-independent fashion. Nevertheless, additional reports have suggested that in addition to the induction of eNOS phosphorylation in response to insulin, insulin also induces an increase in L-arginine transport in endothelial cells, providing higher levels of substrate for NO synthesis [92, 93]. Interestingly, this effect on L-arginine is abrogated by PI3-K/PKB inhibition reinforcing the role of PKB in insulin-dependent modulation of endothelial function. Accordingly, obese and insulin-resistant Zucker rats subjected to L-arginine supplementation exhibited an increase of approximatively 85 % in NO release in comparison with their littermates that were not supplemented with L-arginine [94].
Although enhanced bioavailability of NO is beneficial for improving endothelial functions, excessive production of NO via the activity of iNOS has been suggested to be deleterious in the vasculature [95–97]. In fact, heightened levels of iNOS expression are observed in obese and diabetic states and account for aberrant PI3-K/PKB signalling, insulin resistance and impaired endothelial function [98–100]. Thus, in insulin-resistant states, increased iNOS expression could be an additional mechanism underlying endothelial dysfunction in diabetes or obesity.
26.6 PKC and NO System
Hyperglycaemia and ROS have been shown to activate protein kinase C (PKC) isoforms mainly via diacylglycerol formation [101–103]. Studies have suggested that the PKC isoforms mediate vascular complications by directly activating NADPH oxidase leading to higher ROS generation. Recent reports have shown that activation of PKC-β in endothelial cells blunts PKB phosphorylation in response to insulin and vascular endothelial growth factor [104]. Studies in insulin-resistant Zucker fatty rats reported that inhibition of PKC-β protects against endothelial insulin resistance by preserving insulin-mediated PKB phosphorylation and subsequent NO release [104]. In addition to their modulatory role in the insulin/NO pathway, PKC-β isoforms have also been demonstrated to cause an inhibitory phosphorylation of eNOS on Thr495/497. By maintaining a sustained phosphorylation of eNOS on Thr495/497, PKC-β antagonises agonist-induced eNOS activation and subsequent NO release [105, 106]. Consistent with that, an increase in NO bioavailability was observed following PKC inactivation in endothelial cells [107]. Furthermore, eNOS uncoupling towards ROS generation has recently been shown to be stimulated by PKC via Thr497 phosphorylation of eNOS in endothelial cells reinforcing the role of PKC in the impairment of endothelial function by modulating NO bioavailability [108].
The evidence of the pro-atherogenicity of PKC-β is reinforced by studies where atherosclerosis in ApoE−/−mice is significantly decreased by genetic deletion or ruboxistaurin-mediated pharmacological blockade of PKC-β [109, 110]. PKC-β mediates atherosclerosis via the induction of the early growth response gene (EGR-1) and the matrix metalloproteinase-2. Blockade of PKC-β also reduces the lesion size by lowering the levels of pro-inflammatory proteins and macrophages [109, 110].
26.7 Dyslipidaemia and Regulation of NO System
Dyslipidaemia is associated with vascular endothelial dysfunction and has been suggested as a contributing factor in the pathogenesis of atherosclerosis. Free fatty acids (FFA) are produced from the metabolism of triglycerides and were reported to inhibit insulin-induced NO production resulting in impaired vasodilation [111]. It was found that hypertriglyceridaemia directly impairs endothelial function by stimulating the production of endogenous ADMA known to antagonise NO synthesis by competing with L-arginine [112]. Accordingly, clinical studies reported a decrease in endothelial NO release in obese patients exhibiting elevated plasma levels of ADMA [113, 114]. High levels of FFA can also affect NO bioavailability by impairing PI3-K/PKB/eNOS route in endothelial cells [115, 116]. Additionally, in response to FFA, induction of iNOS production and pro-inflammatory markers has been reported to disrupt NO system resulting in endothelial dysfunction [117, 118]. Moreover, calcium and its downstream effector proteins are potent activators of PKB/eNOS/NO pathway [119], and an attenuation of calcium signalling in endothelial cells exposed to FFA has been reported [120]. Consequently, FFA overload in endothelial cells contributes to eNOS uncoupling by altering calcium-induced eNOS activation [120]. Thus, in the dyslipidaemic state, both altered substrate availability and impaired PI3-K/PKB/eNOS signalling can alter NO bioavailability and contribute to the progression of atherosclerotic vascular disease (Fig. 26.2).
26.8 Concluding Remarks
NO is a well-studied vasoprotective agent and has been suggested to play a protective role in vascular disease by antagonising the physiological and pathological effects of growth factors and vasoactive peptides whose levels are enhanced in vascular diseases. Thus, a reduction in NO bioavailability via substrate or cofactor modulation may be one of the mechanisms that could contribute to the pathogenesis of atherosclerosis. In addition, eNOS activity can be modulated by post-translational phosphorylation events catalysed by protein kinases such as PKB and PKC. The activity of these kinases can be modified by ROS, vasoactive peptides and growth factors during the pathogenesis of atherosclerosis. Currently, efforts are being directed to investigate whether improved availability of eNOS substrate/cofactor and/or modulation of protein kinase signalling pathways could serve as potential therapeutic approaches in the treatment of atherosclerosis.
Acknowledgements
The work in the author’s laboratory is supported by funding from the Canadian Institutes of Health Research (CIHR) operating grant number 67037 to AKS. ERSC was a recipient of a studentship from the Faculty of Graduate and Postdoctoral Studies of the University of Montreal.
References
1.
Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357(Pt 3):593–615.PubMedCentralPubMed
2.
Buchwalow IB, Podzuweit T, Bocker W, Samoilova VE, Thomas S, Wellner M, et al. Vascular smooth muscle and nitric oxide synthase. FASEB J. 2002;16(6):500–8.PubMed
3.
Park CS, Park R, Krishna G. Constitutive expression and structural diversity of inducible isoform of nitric oxide synthase in human tissues. Life Sci. 1996;59(3):219–25.PubMed
4.
Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–76.PubMed
5.
Forstermann U, Boissel JP, Kleinert H. Expressional control of the ‘constitutive’ isoforms of nitric oxide synthase (NOS I and NOS III). FASEB J. 1998;12(10):773–90.PubMed
6.
Nakata S, Tsutsui M, Shimokawa H, Tamura M, Tasaki H, Morishita T, et al. Vascular neuronal NO synthase is selectively upregulated by platelet-derived growth factor: involvement of the MEK/ERK pathway. Arterioscler Thromb Vasc Biol. 2005;25(12):2502–8.PubMed
7.
Nakata S, Tsutsui M, Shimokawa H, Yamashita T, Tanimoto A, Tasaki H, et al. Statin treatment upregulates vascular neuronal nitric oxide synthase through Akt/NF-kappaB pathway. Arterioscler Thromb Vasc Biol. 2007;27(1):92–8.PubMed
8.
Tsutsui M. Neuronal nitric oxide synthase as a novel anti-atherogenic factor. J Atheroscler Thromb. 2004;11(2):41–8.PubMed
9.
Ignarro LJ. Biosynthesis and metabolism of endothelium-derived nitric-oxide. Annu Rev Pharmacol Toxicol. 1990;30:535–60.PubMed
10.
Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279(35):36167–70.PubMed
11.
Ignarro LJ, Degnan JN, Baricos WH, Kadowitz PJ, Wolin MS. Activation of purified guanylate cyclase by nitric oxide requires heme. Comparison of heme-deficient, heme-reconstituted and heme-containing forms of soluble enzyme from bovine lung. Biochim Biophys Acta. 1982;718(1):49–59.PubMed
13.
Krumenacker JS, Hanafy KA, Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res Bull. 2004;62(6):505–15.PubMed
14.
Pfeifer A, Ruth P, Dostmann W, Sausbier M, Klatt P, Hofmann F. Structure and function of cGMP-dependent protein kinases. Rev Physiol Biochem Pharmacol. 1999;135:105–49.PubMed
15.
Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. 2003;93(10):907–16.PubMed
16.
Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci. 2006;11:356–67.PubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


