Fig. 11.1
Neural interface approach to stimulate the vagal nerve. While monitoring heart rate through an implantable transmitter, a remote control system adjusted the intensity of electrical pulses of an implantable miniature radio-controlled electrical stimulator (Reproduced with permission from Li et al. [31])
Shortly after, the group of Hani Sabbah evaluated the effects of cervical VNS in a canine model of intracoronary microembolisation-induced HF [33]. First (2005), they demonstrated that LV volumes and function significantly improved after 3 months of monotherapy with VNS compared with sham-operated animals. The VNS system used (CardioFit, BioControl) had a feedback HR control set to reduce basal HR by approximately 10 %. They also assessed mRNA and protein expression of TNF-α, IL-6, NOS isoforms and connexin-43 (Cx43) in LV tissue. Long-term therapy with VNS significantly decreased both TNF-α and IL-6, tended to normalise the expression of NOS isoforms in the failing LV myocardium and was associated with a major increase in the expression of Cx43. This is a protein-forming gap junctions which is reduced or redistributed from intercalated discs to lateral cell borders in HF [34], increasing susceptibility to arrhythmia in failing hearts [35]. Finally, histology of the right vagus nerve showed normal fascicles and cellular structure when compared to the left vagus. Subsequently (2007), the same group demonstrated that a 3-month combination therapy of VNS and beta-blockade (metoprolol) improved LV haemodynamics beyond what seen with beta-blockade alone (+9.8 ± 0.6 % vs. +5.5 ± 1.2 %). More recently [36], they performed a crossover study using a different VNS system (Boston Scientific Corporation) specifically set with a low stimulation intensity, not affecting HR. Twenty-six canines were enrolled in VNS monotherapy vs control or a crossover study, with crossover occurring at 3 months. Not only in the 6-month stimulated group but also in the crossover study VNS resulted in a significant benefit in LV parameters. Moreover, VNS led to improvement of the plasmatic and tissual biomarkers assessed, including NT-proBNP, proANP and multiple inflammatory molecules (TNF-α, IL-6, BCL-2, caspase-3, Cx43 and the 3 isoforms of NO synthase).
Finally, Zhang et al. [37] studied the effects of chronic VNS in a different canine model of HF, namely, high-rate ventricular pacing. All dogs underwent 8 weeks of high-rate ventricular pacing with concomitant VNS (at an intensity reducing sinus heart rate ≈20 bpm) in the active group and no stimulation in the sham control group. Also, in this study, the treated group showed meaningful benefits in LV haemodynamics, plasmatic levels of inflammatory biomarkers (CRP) and neurohormones (norepinephrine and angiotensin II) despite the fact that HR was not allowed to change due to ventricular pacing. Furthermore, heart rate variability and baroreflex sensitivity, two important markers of autonomic dysfunction in HF, were both significantly improved in VNS dogs.
11.3 Mechanism of Action of Vagal Stimulation
Several mechanisms may contribute to the beneficial effects of vagal stimulation.
Heart rate may decrease acutely during stimulation, albeit minimally if the intensity of stimulation is low and chronically if the autonomic balance is shifted to a less-marked sympathetic dominance. An anti-sympathetic effect is exerted at several levels: centrally because of afferent vagal stimulation and peripherally at both pre- and postsynaptic levels.
Vagal stimulation produces anti-apoptotic effects, facilitates release of NO and has been demonstrated to exert anti-inflammatory effects. The overall mechanisms of action of vagal stimulation are discussed in recent reviews [1, 38–40].
This paper will focus on what we believe is the most intriguing mechanism, namely, the anti-inflammatory effect.
11.4 Inflammation in Heart Failure
Following the original observation by Levine et al. in 1990 of an enhanced inflammatory response associated with HF [41], numerous studies provided evidence of the relationship between inflammation and the severity of HF, its progression and outcome. Notably, inflammatory mediators have been proposed as markers of HF presence, severity or prognosis [42].
There are currently two main recognised inflammatory mechanisms involved in HF: the release of pro-inflammatory cytokines and soluble factors by immune cells, and the dysregulation of nitric oxide synthase in the heart.
The main cytokines up-regulated in HF are the TNF superfamily; the IL-1 family, including IL-1β, IL-18 and IL-6; and a wide range of factors such as s-ICAM-1 or HMGB1 [43]. These mediators have been recognised as pivotal contributors to the development of the main features of HF by a wide range of studies, conducted either in experimental models or in man. These studies showed that high cytokine levels correlate with impaired systolic function and low ejection fraction in experimental models of HF [44–46]. Moreover, pro-inflammatory cytokines negatively influence left ventricular remodelling by activating different biochemical mediators, promoting cardiomyocyte hypertrophy [47] and apoptosis [48], as well as extracellular matrix degradation [49], with the net effect of causing left ventricular dilation [49]. Beside these effects, pro-inflammatory cytokines also cause endothelial dysfunction [50] and abnormalities in the cellular metabolism, as well as the systemic syndrome known as cardiac cachexia, typical of end-stage HF [51]. However, these cytokines do not exert solely deleterious effects, since some of their actions appear to be cardioprotective. For instance, the presence of TNF and IL-6 contributes to limit apoptosis and promote cardiac repair after myocardial ischaemia [52].
The second important inflammatory mechanism associated with HF is the dysregulation of the signalling molecule NO (nitric oxide). NO is synthesised in the heart by three different isoforms of NO synthase (NOS): neuronal NOS (NOS1), endothelial NOS (NOS3) and inducible NOS (NOS2). NOS1 and NOS3 are constitutively expressed in the normal heart, whereas NOS2 is produced in response to TNF and IL-6 in conditions such as in heart failure. NOS1 and NOS3 produce physiological amounts of NO that protect the myocardium from adrenergic stimuli and wall stress, preventing arrhythmias and remodelling. Conversely, NOS2 produces excessive amounts of NO that result in myocardial dysfunction and decreased response to catecholamines [53–55]. Heart failure is characterised by an excess of NO and the coexistence of reactive oxygen species (ROS) and NO. This occurs because of alterations in NOS isoforms produced by the inflammatory process. Namely, NOS2 is up-regulated by TNF, whereas NOS1 is downregulated. This particular pattern produces an increase of myocardial concentrations of NO, playing a role in the transition from adaptative to maladaptative remodelling [43].
11.5 Anti-cytokine Therapies
The growing knowledge of the role of inflammation in heart failure led to studies aimed at reducing this inflammatory response and its consequences. Drugs antagonising cytokines were investigated starting with TNF due to its central role in the inflammatory process associated with heart failure. Indeed, TNF levels are strongly associated with heart failure and its severity [56, 57]. Also, encouraging results were found in in vitro tests [58] and preclinical studies [59] with the anti-TNF agent etanercept. However, the results of the clinical studies with either etanercept [60] or the anti-TNF antibody infliximab [61] were very discouraging since these agents were ineffective and even worsened HF in some cases.
Although targeting inflammation still remains a promising strategy to positively influence the natural history of HF, the failure of anti-cytokine-based trials suggests that the complexity of the cytokine network requires a less simplistic approach. Interfering with a single cytokine such as TNF may eliminate its negative effects but without preserving the potentially beneficial ones. Indeed, there is growing evidence that cytokines classically considered pro-inflammatory exert also anti-inflammatory effects. A better strategy could be the modulation, rather than the suppression, of cytokine production since a balanced immune response is necessary to respond to injury and restore health, in agreement with the cytokine theory of diseases [62].
11.6 The Cholinergic Anti-inflammatory Pathway
At the beginning of this century, the revolutionary hypothesis emerged that the immune activation could be modulated by the nervous system through parasympathetic activity. Tracey proposed that the autonomic nervous system could influence inflammation, in a way similar to its influences on the inner splanchno-thoracic organs, through unconscious reflex pathways that maintain the fine homeostasis of the immune system [63]. This so-called inflammatory reflex can sense inflammation through vagal afferents and responds with an increase of vagal efferent activity. This cholinergic anti-inflammatory pathway can inhibit further release of pro-inflammatory mediators through the action of acetylcholine on macrophages and other immune cells.
The existence of this pathway was first suggested in 2000 by Borovikova et al. [64] who showed that electrical stimulation of the vagal nerve in endotoxemic mice inhibits the macrophagic release of TNF and other pro-inflammatory cytokines, without affecting the production of anti-inflammatory cytokines, as IL-10, thus preventing the development of shock. Few years later, Pavlov and colleagues demonstrated that intrathecal administration of muscarinic agonists in endotoxemic rats increases vagal outflow, provoking a rise of instantaneous heart rate variability (HRV) and a drop of pro-inflammatory cytokine levels [65]. Further evidence of the activity of the cholinergic anti-inflammatory pathway was found not only in sepsis but also in experimental models of various diseases such as haemorrhagic shock [66], rheumatoid arthritis [67], pancreatitis [68] and ischaemia and reperfusion of the lung [69] and heart [70, 71].
11.7 Physiology of the Cholinergic Anti-inflammatory Pathway
As every visceral reflex, the cholinergic anti-inflammatory pathway is made up by an afferent arc providing input to a central integrative point and an efferent arc acting on the target cells to modify their activity (Fig. 11.2). The afferent arc is constituted of vagal sensitive neurons that fire in response to the presence of IL-1, TNF and other inflammatory cytokines, detected by glomic cells situated in most organs and tissues. These sensitive neurons project to the hypothalamus and the brainstem, in the nucleus of the solitary tract. In the brainstem, a neural network processes the information and regulates the outflow from vagal nuclei such as the nucleus ambiguous and the dorsal motor nucleus where the efferent vagal neurons are located. These vagal fibres release acetylcholine in the inner organs and in the spleen, the major repository of pro-inflammatory monocytes and macrophages. Acetylcholine binds the α7 subunity of the nicotinic receptor, situated on the surface of macrophages, endothelial cells and B and T cells. The activation of the receptor generates inhibition of the production of pro-inflammatory cytokines, as TNF, IL-1, IL-6 and IL-8, without influencing the production of anti-inflammatory cytokines, such as IL-10. Efferents to the adrenal gland favour the production of corticosteroids, providing an additional anti-inflammatory mechanism [72, 73].
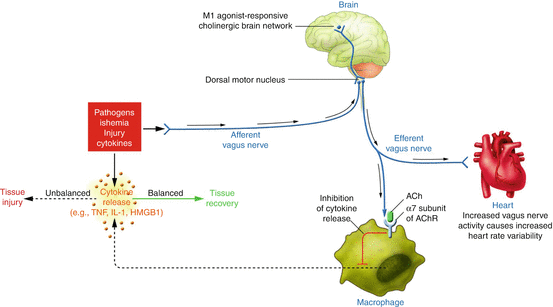
Fig. 11.2
Cholinergic anti-inflammatory pathway balances cytokine production. Several forms of injury lead to inflammatory response with cytokines release. If this reaction is excessive, these mediators can cause disease. The cholinergic anti-inflammatory pathway inhibits cytokine production through the key receptor alpha7-nicotinic AChR expressed on macrophages, polymorphonuclear cells, endothelium and other cells. Vagal fibres also exert favourable autonomic modulation at cardiac level (Reproduced with permission from Tracey [72])
11.8 Cholinergic Anti-inflammatory Pathway as Therapeutic Target
The cholinergic anti-inflammatory pathway may provide the possibility to prevent the deleterious effects of an excessive inflammatory and immune response without suppressing it completely.
In a rat model of ischaemia and reperfusion injury (widely accepted as an inflammatory process) obtained by suprarenal aortic clamping, Bernik et al. showed that vagal stimulation attenuated TNF levels in liver, serum and heart and reduced hypotension and shock [74]. In a mouse model of laparotomy and intestinal manipulation, de Jonge et al. showed that the anti-inflammatory action of alpha-7nAChR activation in peritoneal macrophages was associated with activation of the Jak2/Stat3 signalling pathway, resulting in inhibition of the synthesis of pro-inflammatory cytokines [75]. Interestingly, the same Jak/Stat pathway was found to be implicated in the early phase of ischaemic preconditioning: cardioprotection provided by preconditioning was blocked by AG490 which inhibited Jak2/Stat3 phosphorylation [76, 77]. Furthermore, Dawn et al. showed that both IL-6 and TNF-α are involved in the development of ischaemic preconditioning; specifically, TNF seems to act via the activation of NF-kB and AP1 transcription factors [78, 79]. These findings appear in agreement with the concept that remote conditioning is likely to confer its protective effects through efferent vagal activity [80]. A key mechanism leading to reperfusion injury is the prolonged opening of a nonspecific pore in the inner mitochondrial membrane (permeability transition pore, PTP) leading to ATP depletion, activation of death messengers such as caspases and ultimately to cell death [81]. In a murine model of ischaemia/reperfusion associated with fixed rate pacing [82], vagal stimulation improved LV systolic and diastolic pressures, prevented downregulation of the anti-apoptotic protein Bcl-2 and suppressed caspase-3 activation. These favourable effects were abolished by a PTP opener, atractyloside. These findings are in agreement with the recent demonstration that the cholinergic nicotinic anti-inflammatory pathway is the main mechanism of protection in case of remote vascular injury after cardiac ischemia and reperfusion. Indeed vagal stimulation not only improved cardiac function but also up-regulated M3AChR and alpha7nAChR and ameliorated vascular function and degradation of endothelial structure in mesenteric arteries [83].
Additionally, it was recently reported that vagal nerve stimulation decreases the inflammatory response to haemorrhage and improves coagulation in a model of haemorrhagic shock [84]. Also, Schulte et al. showed that vagal activity may play a major role in maintaining haemodynamic stability and cardiac immune homeostasis during septic shock in a model of systemic inflammation [85].
Overall, this new scenario led investigators to evaluate the potential of a pharmacological approach to enhance vagal activity thereby activating the cholinergic anti-inflammatory pathway without using electrical devices. A promising agent appears GTS-21, a selective alpha7-nicotinic acetylcholine receptor agonist. In a murine model of endotoxemia and severe sepsis, GTS-21 inhibited TNF, HMGB-1 and NF-kB and improved survival [86]. Recently, Kox et al. reported an anti-inflammatory effect of this agent on the innate immune response during experimental human endotoxemia [87]. Oral administration of GTS-21 was found to be safe in human volunteers and, although no significant differences in inflammatory markers were found between subjects in the active and placebo arm, within the GTS-21-treated group, higher plasma concentrations correlated with lower levels of TNF-α, IL-6 and IL-1RA after intravenous injection of 2 ng/kg Escherichia coli-derived lipopolysaccharides.
Several years ago GTS-21 was found to be able to attenuate the impairment of spatial cognitive deficit and the progressive neuronal degeneration induced by permanent occlusion of the bilateral common carotid arteries in rats [88], thus suggesting that this compound may be beneficial for the treatment of neurodegenerative diseases following chronic cerebral hypoperfusion. Also, pretreatment with a cholinergic agonist, nicotine or GTS-21 significantly attenuated both renal dysfunction and tubular necrosis induced by experimental renal ischaemia in rats [89].
11.9 Cholinergic Anti-inflammatory Pathway and Inflammation in Heart Failure
Given the fact that vagal nerve stimulation showed a remarkable ability to modulate innate immune responses in various inflammatory diseases, it would appear logical to assess the potential of vagal stimulation to normalise the inflammatory status associated with heart failure.
The main theoretical advantage in stimulating vagus nerve instead of inhibiting inflammation by pharmacological intervention is that the cholinergic pathway is an already existing physiological anti-inflammatory mechanism which may be simply reinforced or fine-tuned. This is particularly attractive after the failure of pharmacological interventions targeting the inflammatory responses in heart failure [60].
Very relevant appears the recent study by Ismahil and colleagues [90] highlighting the role of monocytes/macrophages and dendritic cells in chronic heart failure. In a murine experimental model of heart failure, pro-inflammatory macrophages and monocytes were found to be increased both in the heart and in blood but decreased in the spleen which, undergoes marked remodelling in HF. The authors demonstrated that the spleen is essential for the progression of remodelling and inflammation in HF since splenectomy reversed inflammation and remodelling in mice with HF, whereas splenocytes from mice with HF to injected in recipient healthy mice homed in the heart and eventually caused the typical symptoms of HF.
11.10 Clinical Studies with Vagal Stimulation
Clinically, chronic vagal stimulation (VS) has been used for the management of drug-refractory epilepsy [91] and, more recently, depression [92].
Three clinical studies of VS in patients with systolic HF have been reported so far. Table 11.1 summarises the main baseline characteristics of the patients enrolled. The CardioFit pilot study – an extension of the first-in-man single-centre feasibility study [93] – used the CardioFit 5000 device (BioControl Medical, Yehud, Israel), a system including, in addition to a right vagal nerve electrode, a right ventricular sensing lead allowing VS to be synchronised to the QRS complex and withheld in case of low heart rate. This open-label phase II study [94] enrolled 32 patients in total (94 % men; mean age 56 ± 11 years) with a history of chronic NYHA class II–IV HF and an LVEF of 23 ± 8 %. All patients were receiving optimal medical therapy (OMT) and 19 patients had an implantable cardioverter-defibrillator (ICD). Vagal stimulation intensity was mostly limited by patient’s symptoms and was uptitrated to 4.1 ± 1.2 mA (with 1–2 pulses per cardiac cycle). The heart rate was minimally affected acutely during stimulation but decreased significantly during the study from 82 ± 13 to 76 ± 13 b/min. After a follow-up of 6 months, 59 % of patients improved by at least one NYHA class and both the distance on the 6-min walk test and the Minnesota Living with Heart Failure Questionnaire quality-of-life score improved significantly. Blinded echocardiography revealed a non-significant decrease in LV end-diastolic volume index, a significant reduction in LV end-systolic volume index and a significant increase in LVEF (from 22 ± 7 % to 29 ± 8 %). Follow-up in a group of patients at 1 and 2 years showed persistence of the increase in LVEF.
Table 11.1
Patients’ characteristics at baseline in the three published VS clinical studies
Parameter | CardioFit | ANTHEM-HF | NECTAR-HF |
|---|---|---|---|
Patients number | 32 | 60 | 96 (87 paired data) |
Age (years) | 56 ± 11 | 51 ± 12 | 59 ± 11 |
NYHA class II/III/IV | 15/15/2 | 34/26/0 | 14/73/0 |
Ischaemic HF (%) | 62 | 75 | 67 |
Patients taking BB (%) | 97 % | 100 % | 94 % |
Patients taking ACE-i or ARB (%) | 97 % | 85 % | ACE-i 78 %, ARB 25 % |
Patients taking MRA (%) | 97 % | 75 % | 70 |
LVEF baseline (%) | 23 ± 8 | 32 ± 7 | 30 ± 6 |
LVESV baseline (ml) | 185 ± 63 | 108 ± 40 | 155 ± 58 |
ICD/CRT-D/no device | 19/0/13 | 0/0/60 | 73/9/13 |
A VS system with no RV sensing lead was used in the Autonomic Regulation Therapy via Left or Right Cervical Vagus Nerve Stimulation in Patients with Chronic Heart Failure (ANTHEM-HF) study [95], an open-label phase II trial that enrolled 60 patients with NYHA class II and III HF, LVEF <40 % and QRS <150 ms. Patients were randomised to either left or right cervical VS uptitrating the amplitude over a 10-week period and reaching an average value of 2.0 ± 0.6 mA; then, they were followed for 6 months. Procedure and device-related complications appeared low. In the pooled right + left VS analysis, LVEF increased by 4.5 % (p < 0.05), whereas the co-primary efficacy end point, LV end-systolic volume, decreased non-significantly by 4.1 ml. A non-significant trend towards greater improvement with right VS was observed. There was a significant improvement in the Minnesota Living with Heart Failure score and 77 % of patients improved by at least one NYHA.
The Neural Cardiac Therapy for Heart Failure (NECTAR-HF) study was a phase II study which enrolled 96 patients with NYHA class II and III HF, an LVEF <35 %, a QRS <130 ms and LV end-diastolic diameter >55 mm. At variance with the previous study, all patients received a right-sided device implant and were randomised 2:1 to active treatment or sham treatment (with the device on only during the titration visits) for the first 6 months; thereafter, all patients received active treatment. The device used in NECTAR-HF was similar to that used in ANTHEM, also lacking an RV lead [96]. The stimulation intensity was 1.3 ± 0.8 mA. There were no major safety concerns; an assessment of blinding performed at 6 months revealed that 70 % of the patients assigned to active treatment correctly guessed their randomisation group. There was no effect on primary efficacy end point, which was the change in LV end-systolic diameter at 6-month follow-up [97]. Several secondary end points, including LV end-diastolic dimension, LV end-systolic volume, LVEF, peak V02, and N-terminal pro-hormone brain natriuretic peptide, were also not different between groups. On the other hand, statistically significant improvements were found in the active therapy group in quality of life for the Minnesota Living with Heart Failure Questionnaire (p < 0.049) and the New York Heart Association class (p < 0.032).
Overall, the three studies yielded different results. Although several factors may account for this discrepancy, two possibilities deserve to be mentioned. First, only one trial was controlled and (albeit partially) blinded and it was the one with neutral findings (NECTAR-HF). Second, the amplitude reached by vagal stimulation was very different in the three studies. Lower frequencies of stimulation allow greater amplitudes to be reached with tolerable side effects. Accordingly, the stimulation intensity varied in the three studies: it was 1.3 ± 08 mA in NECTAR-HF (range 0.3–3.5), stimulating at 20 Hz; 2.0 ± 0.6 mA in ANTHEM (maximum amplitude allowed 3 mA), stimulating at 10 Hz; and 4.2 ± 1.2 mA (range 1.1–5.5) in the CardioFit pilot trial, stimulating at 1–2 Hz. Higher amplitudes allow a greater recruitment of nerve fibres. Experimental studies in conscious dogs have shown that amplitudes such as those used in the NECTAR-HF study, albeit providing an improvement in LV function [36], recruit only a minority of the fibres in the cervical vagal trunk [98]. Thus, it is likely that the lower amplitude of VS in NECTAR-HF and ANTHEM recruited an insufficient number of vagal fibres and that this could have played a major role in the different results.
Hopefully, the picture will be more clear when the results of the ongoing Increase of Vagal Tone in Heart Failure (INOVATE-HF) study [99] will be available (likely in 2017). This is an international, multicentre randomised clinical trial assessing safety and efficacy of the CardioFit™ vagal stimulation system in patients with symptomatic HF who are on optimal medical treatment [99]. INOVATE-HF is randomising 650 patients with NYHA class III symptoms, an LVEF ≤40 % and LV end-diastolic dimensions 50–80 mm in a 3:2 ratio to either active treatment (implanted) or continuation of medical therapy (not implanted). The primary end point of the study is the composite of all-cause mortality or unplanned HF hospitalisation equivalent, using a time to first event analysis.
11.11 Conclusions
Vagal nerve stimulation is a promising new approach for patients with chronic heart failure whose clinical usefulness needs to be assessed in large randomised clinical trials. Activation of the cholinergic anti-inflammatory pathway is likely to play an important role in the favourable effects of vagal stimulation.
Conflict of Interest Disclosures
GMDF is a consultant for Boston Scientific; PJS for BioControl Medical Ltd.
References
1.
Floras JS. Sympathetic nervous system activation in human heart failure: clinical implications of an updated model. J Am Coll Cardiol. 2009;54:375–85.PubMedCrossRef
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


