The objective of the present study was to assess the safety and effectiveness of vernakalant hydrochloride injection (RSD1235), a novel antiarrhythmic drug, for the conversion of atrial fibrillation (AF) or atrial flutter to sinus rhythm (SR). Patients with either AF or atrial flutter were randomized in a 1:1 ratio to receive vernakalant (n = 138) or placebo (n = 138) and were stratified by an arrhythmia duration of >3 hours to ≤7 days (short duration) and 8 to ≤45 days (long duration). The first infusion of placebo or vernakalant (3 mg/kg) was given for 10 minutes followed by a second infusion of placebo or vernakalant (2 mg/kg) 15 minutes later if the arrhythmia had not terminated. A total of 265 patients were randomized and received treatment. The primary end point was conversion of AF to SR for ≥1 minute within 90 minutes of the start of the drug infusion in the short-duration AF group. Of the 86 patients receiving vernakalant in the short-duration AF group, 44 (51.2%) demonstrated conversion to SR compared to 3 (3.6%) of the 84 in the placebo group (p <0.0001). The median interval to conversion of short-duration AF to SR in the responders given vernakalant was 8 minutes. Of the entire AF population (short- and long-duration AF), 47 (39.8%) of the 118 vernakalant patients experienced conversion of AF to SR compared to 4 (3.3%) of the 121 placebo patients (p <0.0001). Transient dysgeusia and sneezing were the most common adverse events in the vernakalant patients. One vernakalant patient who had severe aortic stenosis experienced hypotension and ventricular fibrillation and died. In conclusion, vernakalant demonstrated a rapid and high rate of conversion for short-duration AF and was well tolerated.
Vernakalant hydrochloride (RSD1235) is being developed as an intravenous drug to pharmacologically convert atrial fibrillation (AF) to sinus rhythm (SR). Vernakalant acts on the heart by blocking the K + channels (IK ur , I to , IK r , and IK ACh ) in a manner that predominantly affects atrial repolarization, combined with a concentration-, voltage-, and frequency-dependent blockade of Na + channels, with no effect on Ca 2+ channels. In animal models and an electrophysiologic study in humans, vernakalant prolonged the atrial refractory period without affecting the ventricular refractoriness. Previous trials have demonstrated that intravenous vernakalant effectively converts AF to SR. The present trial is the second of 2 pivotal trials forming the basis for regulatory approval by the United States Food and Drug Administration. An important difference from the first pivotal trial (Atrial Arrhythmia Conversion Trial [ACT]-I) is that patients with atrial flutter (AFL) were also included in the present study.
Methods
The present study was a prospective, randomized, double-blind, placebo-controlled international trial conducted from June 27, 2004 to August 1, 2005. The institutional or regional review board at each site approved the protocol, and the patients gave written informed consent before starting the study procedures. An unblinded independent data safety monitoring board was used throughout the trial. The sponsors and members of the steering committee had full access to the data. The sponsor performed the initial data analysis, which was reviewed by the steering committee, who mandated additional analyses.
To be eligible, patients had to have had sustained AF or AFL for >3 hours but ≤45 days. Patients were excluded if their baseline characteristics were not within these boundaries: age ≥18 years, body weight 45 to 136 kg (99 to 300 lb), adequate anticoagulation, and systolic blood pressure >90 and <160 mm Hg and diastolic blood pressure <95 mm Hg. Other key exclusions included QRS >0.14 seconds without a pacemaker, a ventricular rate of <50 beats/min without a pacemaker, an uncorrected QT interval of >0.440 seconds, class IV heart failure, acute coronary syndrome, and myocardial infarction or cardiac surgery within 30 days before randomization. The protocol was amended on March 24, 2005 to include severe valvular stenosis, hypertrophic obstructive cardiomyopathy, restrictive cardiomyopathy, or constrictive pericarditis as exclusion criteria. Other standard exclusions have been detailed in the study protocol (Cardiome Pharma, Vancouver, British Columbia, Canada, protocol 1235-0504; and Fujisawa Healthcare, North Deerfield, Illinois, protocol 04-7-010; Clinicaltrials.gov number, NCT00115791 , March 31, 2004). β-Adrenergic blockers, calcium channel blockers, or digoxin could be used for control of the ventricular rate for ≤2 hours before study drug infusion. Patients were ineligible if they had received intravenous class I or III antiarrhythmics, including amiodarone, within 24 hours before study drug infusion.
The study design is depicted in Figure 1 . Patients received either a 10-minute infusion of vernakalant (3 mg/kg) or placebo, followed by a 15-minute observation period. If the patient was still in AF or AFL, an additional 10-minute infusion of vernakalant (2 mg/kg) or placebo was administered. The infusions were to be discontinued if the QT interval increased to ≥0.550 seconds or increased by >25% from baseline or if the heart rate decreased ≥40 beats/min for ≥30 seconds and was associated with symptoms of bradycardia or to <40 beats/min for ≥30 seconds, regardless of symptoms. Additional reasons for study infusion discontinuation have been provided in the study protocol ( NCT00115791 ). Patients were observed in the hospital for a minimum of 8 hours after the study drug infusion. In addition to continuous telemetry for the first 2 hours, electrocardiograms were recorded and vital signs were measured at screening, baseline, every 5 minutes from the start of infusion to 50 minutes after the start of infusion, at 90 minutes and 2, 4, 8, and 24 hours, on conversion to SR or at the occurrence of a serious adverse event (AE), and at 1 week after dosing. In addition to telemetry, a Holter recording was obtained from screening to 24 hours after dosing. The protocol included a clinical events committee and a core electrocardiogram laboratory.
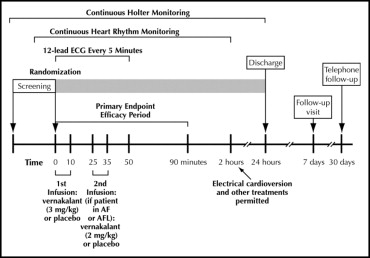
The primary efficacy end point was the proportion of patients in the short-duration AF group who had conversion to SR for ≥1 minute within 90 minutes of drug initiation. Patients demonstrating conversion to SR within 90 minutes of dosing were categorized as responders. The secondary efficacy end points included the interval to conversion of short-duration AF to SR. Exploratory efficacy end points have been included in the study protocol ( NCT00115791 ).
The efficacy and safety analyses are all presented as modified intention-to-treat (all patients who received the study drug). The analyses based on an intention-to-treat approach were consistent with the results reported here. The baseline characteristics were compared between groups using a one-way analysis of variance with a fixed effect for treatment for continuous variables and a chi-square test or Fisher’s exact test for categorical variables. The primary end point was analyzed using the Cochran-Mantel-Haenszel test stratified by the center; this test was also used in the analysis of the group of patients with an AF duration of 3 hours to 45 days. Fisher’s exact test was used in the analysis of the group with an AF duration of 8 to 45 days. The Kaplan-Meier method was used to summarize the interval to conversion, with the log-rank test used to compare the distributions. The sample sizes were based on assumed conversion rates of 25% and 50% in the short-duration group and 5% and 30% in the long-duration group for the placebo and vernakalant groups, respectively. Using a 2-sided chi-square test (p = 0.05), 77 patients in each short-duration group would provide 90% power to detect a 25% difference between the conversion rates with vernakalant and placebo. Using on a 2-sided Fisher exact test (p = 0.05), 33 patients in each long-duration group would provide 70% power to detect a 25% difference in conversion rate between the treatments. The proportion of patients who were in SR at 24 hours after treatment was estimated using life-table methods. Separate analyses of the long-duration AF group and the AFL groups were also prespecified.
Results
The disposition of the 276 study patients is depicted in Figure 2 . No patient had previously participated in a vernakalant study, including ACT I. The baseline demographics and baseline medical therapies are listed in Table 1 . The treatment groups were relatively well-balanced. Of the 86 vernakalant patients in the primary efficacy group, 44 (51.2%) demonstrated conversion to SR for ≥1 minute within 90 minutes of infusion (primary efficacy end point) compared to 3 (3.6%) of the 84 placebo patients (p <0.0001; Figure 3 ). In the long-duration AF group, 1 (2.7%) of 37 patients given placebo versus 3 (9.4%) of 32 patients given vernakalant (p = 0.330) demonstrated conversion of AF to SR within 90 minutes.
| Variable | Short-Duration AF | Long-Duration AF | AFL | |||
|---|---|---|---|---|---|---|
| Placebo (n = 84) | Vernakalant (n = 86) | Placebo (n = 37) | Vernakalant (n = 32) | Placebo (n = 9) | Vernakalant (n = 14) | |
| Men | 57 (68%) | 61 (71%) | 21 (57%) | 20 (62%) | 8 (89%) | 11 (79%) |
| White | 84 (100%) | 86 (100%) | 36 (97%) | 31 (97%) | 8 (89%) | 14 (100%) |
| Age (years) | 60 ± 15 | 60 ± 16 | 66 ± 10 | 65 ± 11 | 60 ± 6 | 61 ± 10 |
| Heart failure | 11 (13%) | 12 (14%) | 11 (30%) | 11 (34%) | 3 (33%) | 4 (29%) |
| Diabetes mellitus | 9 (11%) | 4 (5%) | 7 (19%) | 4 (12%) | 2 (22%) | 2 (14%) |
| Hypertension ⁎ | 28 (33%) | 41 (48%) | 17 (46%) | 16 (50%) | 8 (89%) | 5 (36%) † |
| Hyperlipidemia ⁎ | 21 (25%) | 31 (36%) | 16 (43%) | 13 (41%) | 3 (33%) | 5 (36%) |
| Pacemaker | 3 (4%) | 1 (1%) | 2 (5%) | 2 (6%) | 0 | 0 |
| Ventricular tachycardia | 1 (1%) | 1 (1%) | 2 (5%) | 0 | 0 | 0 |
| Coronary heart disease | 10 (12%) | 5 (6%) | 4 (11%) | 7 (22%) | 2 (22%) | 3 (21%) |
| Myocardial infarction | 4 (5%) | 5 (6%) | 3 (8%) | 2 (6%) | 1 (11%) | 2 (14%) |
| Concomitant therapy | ||||||
| Class I antiarrhythmic drugs ‡ | 6 (7%) | 16 (19%) § | 0 | 1 (3%) | 1 (11%) | 0 |
| Class III antiarrhythmic drugs ‡ | 14 (17%) | 13 (15%) | 8 (22%) | 3 (9%) | 3 (33%) | 5 (36%) |
| β Blockers | 49 (58%) | 52 (60%) | 28 (76%) | 22 (69%) | 6 (67%) | 9 (64%) |
| Calcium channel blockers | 20 (24%) | 16 (19%) | 6 (16%) | 5 (16%) | 6 (67%) | 3 (21%) |
| Digoxin | 13 (16%) | 5 (6%) ¶ | 11 (30%) | 14 (44%) | 3 (33%) | 1 (7%) |
| Baseline atrial fibrillation or atrial flutter symptoms | 71 (84%) | 64 (74%) | 28 (76%) | 27 (84%) | 6 (67%) | 10 (71%) |
⁎ Determined by clinical assessments made by local centers.
† p = 0.029 compared to placebo.
§ p = 0.026 compared to placebo.
¶ p = 0.041 compared to placebo.
‡ Class I antiarrhythmics included disopyramide, flecainide, procainamide, and propafenone; class III antiarrhythmics included amiodarone, ibutilide, and sotalol.
The median interval to conversion to SR among the responders given vernakalant with short-duration AF was 8 minutes ( Figure 4 and Table 2 ). Of the 44 vernakalant patients in the primary efficacy group who demonstrated conversion, 36 (81.8%) did so after the first dose. Of the 44 patients with short-duration AF who had conversion to SR, only 1 patient relapsed to AF within 24 hours ( Table 2 ). At 90 minutes, the percentage of patients with short-duration AF who did not have any AF or AFL symptoms was significantly greater among those given vernakalant than among those given placebo (50.0% vs 28.6%, p = 0.004).
| End Point Description | Placebo | Vernakalant | p Value |
|---|---|---|---|
| Short-duration atrial fibrillation population (n) | 84 | 86 | |
| Conversion to sinus rhythm for ≥1 minute within 90 minutes | 3 (4%) | 44 (52%) | <0.0001 |
| Median time to conversion to sinus rhythm (min) | 34 | 8 | |
| Patients in sinus rhythm at 24 hours after study drug ⁎ | 3 (100%) | 35 (98%) | |
| Patients without atrial fibrillation or atrial flutter symptoms at 90 minutes | 24 (29%) | 43 (50.0%) | 0.004 |
| Long-duration atrial fibrillation population (n) | 37 | 32 | |
| Conversion to sinus rhythm for ≥1 minute within 90 minutes | 1 (3%) | 3 (9%) | 0.33 |
| Median time to conversion to sinus rhythm (min) | 40 | 11 | |
| Patients in sinus rhythm at 24 hours after study drug ⁎ | 1 (100%) | 1 (100%) | |
| Patients without atrial fibrillation or atrial flutter symptoms at 90 minutes | 12 (32%) | 11 (34%) | 1.00 |
| Entire atrial fibrillation population (n) | 121 | 118 | |
| Conversion to sinus rhythm for ≥1 minute within 90 minutes | 4 (3%) | 47 (40%) | <0.0001 |
| Median time to conversion to sinus rhythm (min) | 37 | 9 | |
| Patients in sinus rhythm at 24 hours after study drug ⁎ | 4 (100%) | 36 (98%) | |
| Patients without atrial fibrillation or atrial flutter symptoms at 90 minutes | 36 (30%) | 54 (46%) | 0.011 |
⁎ Life-table estimate; at each point, only patients who demonstrated conversion to SR at previous point were included in calculation; patients with missing data not included.
A total of 15 vernakalant patients with AF at baseline (12.7%) developed new AFL in the first 90 minutes, 12 after the first infusion and 3 after the second. Of these 15 patients, 5 subsequently demonstrated conversion to SR within 90 minutes, and 7 underwent electrical cardioversion. Of the 3 remaining patients, 1 was in SR at hour 4 and 1 at hour 8, and 1 patient remained in AF. None of these AFL episodes were associated with 1:1 atrioventricular conduction.
A total of 23 patients presenting with AFL were enrolled. Of the 14 vernakalant patients, 1 demonstrated conversion to SR compared to none of the 9 placebo patients. Electrical cardioversion was subsequently attempted in 91 (69.5%) of 131 patients given placebo and 54 (40.3%) of 134 patients given vernakalant in the 2 to 24-hour period after infusion in those who had not demonstrated conversion to SR. A median of one shock with a median energy of 200 J was given in each treatment group, with 79 (86.8%) of 91 patients given placebo and 51 (94.4%) of 54 patients given vernakalant achieving successful cardioversion.
Deaths and serious AEs were assessed for ≤30 days after study drug infusion. One death occurred in the trial. A 64-year-old patient with a history of severe aortic stenosis and New York Heart Association class II heart failure presented with acute coronary syndrome and AF with a ventricular rate of ∼150 beats/min. Control of the ventricular response was attempted with intravenous metoprolol. Transient hypotension accompanied these attempts. Despite the hemodynamic instability, the patient was enrolled and randomized to receive vernakalant. He received 2 infusions of vernakalant, becoming hypotensive after each infusion. At minute 47 after the start of the first infusion, he developed ventricular fibrillation followed by electromechanical dissociation, and resuscitation efforts were unsuccessful. Because of this unfortunate case, a subsequent protocol amendment was added that excluded patients with severe valvular stenosis. This AE information was transmitted to all clinical sites, and medical monitors reviewed the screening process at all sites.
In the first 24 hours after infusion, 6 patients (4.6%) given placebo and 3 patients (2.2%) given vernakalant had serious AEs. A total of 17 placebo patients (13.0%) and 14 vernakalant patients (10.4%) had serious AEs reported during the entire 30-day study. Most events were of cardiac origin, with the most common recurrent AF (placebo 3.8% and vernakalant 5.2%). No cases of torsades de pointes developed in the vernakalant patients. One case of torsades de pointes occurred 2 days after study drug infusion in a placebo patient who underwent electrical cardioversion and had received escalating doses of sotalol. One day after receiving the study drug infusion, another patient given placebo had an asymptomatic 6-beat run of ventricular tachycardia at a rate of 152 beats/min.
Ventricular arrhythmia events were assessed using multiple data sources, including AEs, 12-lead electrocardiograms, and 24-hour Holter recordings ( Table 3 ). The analyses were conducted during the 0 to 24-hour period, which was further divided into 0- to 2-hour and 2- to 24-hour periods because other treatments for AF or AFL were allowed after 2 hours. The incidence of ventricular tachycardia at 0 to 2 hours and 2 to 24 hours after dosing was similar between those given vernakalant and those given placebo.



