Fig. 50.1
Original ECG registrations from a patient in SR. Arrows indicate P waves. During normal sinus rhythm, P waves are upright in lead II and V2 and followed 1:1 by an QRS complex
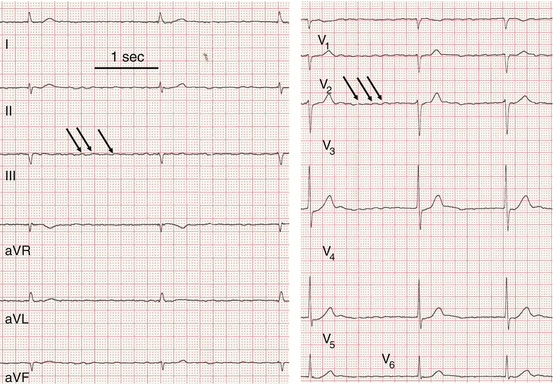
Fig. 50.2
Original ECG registrations from a patient with AF. Arrows indicate P waves are much faster, but smaller and QRS follows irregularly
50.2 Drugs Used for Rate Control
Many patients with a new-onset AF need slowing of the AV conduction (rate control), irrespective of whether restoration of the sinus rhythm will be a therapeutic goal. In patients, where sinus rhythm control is not a suitable treatment option, rate control may be a lifelong intervention. Digitalis glycosides were the first drug class used to treat patients with AF. Digitalis slows down AV-node conduction and thereby reduces the ventricular rate even during fast activation of the atria in AF. The enormous efficacy of this drug, with respect to slowing the heartbeat, is reflected in its name: “opium of the heart.” However, proper dosing has always been a challenge since digitalis glycosides have a rather slow and complex pharmacokinetic in conjunction with a narrow therapeutic window that includes life-threatening side effects [1]. Use of digitalis glycosides is safe when low doses are used and in some cases are controlled by monitoring serum levels of digoxin [2]. However, the vast majority of electrophysiological effects of digitalis glycosides in different regions of the heart give concerns about general safety of that drug class. Prospective data about digitalis efficacy in patients with AF are not available. Recently published large retrospective study with sophisticated statistical methods applied clearly indicates negative outcomes for digitalis treatment in patients with AF [3]. Actual guidelines of AHA/ACC and ESC still recommend digitalis use for rate control in AF. However, irrespective of whether we will have hard prospective data in the future, we have to expect restrictions on the use of digitalis in patients with AF, since two other drug classes with a more favorable safety profile can now be used for rate control.
L-type Ca2+-channels critically regulate conduction in the AV node where Ca2+-channels instead of sodium channels conduct upstroke of action potentials. Two L-type Ca2+-channel blockers, diltiazem and verapamil, are able to provoke marked slowing of ventricular rate during AF [4, 5]. The β-adrenoceptor antagonists slow down AV conduction especially when sympathetic tone is increased as in case of exercise or excitement. Keeping in mind the beneficial effects of β-adrenoceptor antagonists in heart failure, this drug class is widely used in patients with AF and concomitant structural heart disease (~70 % in Europe). There are no obvious differences between different β-adrenoceptor antagonists with respect to slowing down AV conduction (Chaps. 5 and 8).
50.3 Antiarrhythmic Drugs
Atrial flutter is based on larger macro-reentry circuits compared to AF generating clearly distinguishable P waves in the ECG (see Fig. 50.3). Isolated atrial flutter is a rare condition [6]. AF and atrial flutter coexist in many patients. The larger reentry circuits in atrial flutter simplify treatment by catheter ablation and antiarrhythmic drugs. However, AF and atrial flutter probably respond to treatment aimed at prevention of recurrence or improvement of survival and cardiovascular complications in a similar way and can be treated as one entity [7]. Therefore, we do not differentiate between AF and atrial flutter. Classic antiarrhythmic agents can stop arrhythmias. They do so as they have marked effects on the excitability of cardiac tissue. This intervention may be highly effective in acute settings, and even serious side effects may be accepted as long as treatment duration is short and physicians give the drug under controlled conditions.
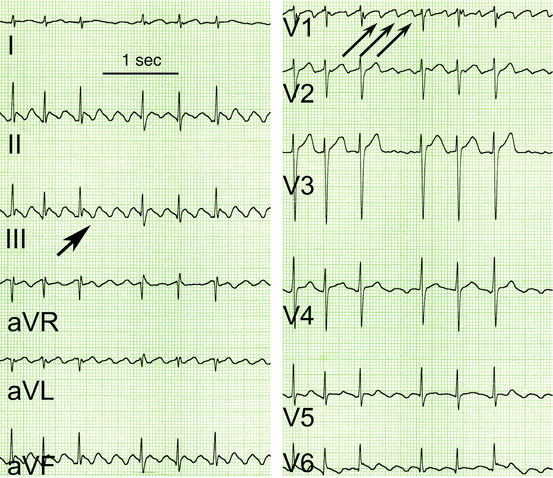
Fig. 50.3
Original ECG registrations from a patient with atrial flutter. Arrows indicate P waves there are much more P waves than QRS complexes. However, P waves are clearly distinguishable and are followed in a 3:1 or 2:1 mode by a QRS complex (best seen in V2)
50.3.1 Class I Drugs
Efficacy of quinine to stop AF was first described in 1759 and introduced as a “pill in the pocket” approach for new-onset AF by a non-medic in 1922 [8]. Quinidine is the d-isomer of quinine, has a higher efficacy than quinine to stop AF, and was therefore used as an antiarrhythmic agent. From a general pharmacology perspective, quinidine shows a wide range of serious adverse effects. Quinidine affects gastrointestinal motility [9], and it should be noted that in the early days of quinidine treatment dosage was increased up to the point when patients started to vomit. Transient thrombocytopenia and hemolytic anemia can occur [10]. Quinidine also blocks α-adrenoceptors [11] and muscarinic receptors [12]. Reflex release of catecholamines upon vasodilation as well as release of inhibitory effects of acetylcholine will increase speed of AV conduction with a harmful increase in ventricular rate during AF. Increased AV conduction is especially harmful in the case of atrial flutter, since ventricular rate can reach critical high values. Therefore, quinidine is often used in conjunction with other drugs that slow AV conduction. Pharmacokinetic properties gave rise to concern on its therapy. Quinidine has high plasma protein binding and can thereby increase serum levels of other drugs bound to plasma proteins [13]. Cytochrome P450 enzymes extensively metabolize quinidine, and many of its metabolites show activity. Most importantly, quinidine can provoke life-threatening ventricular arrhythmias: torsades de pointes. The first ventricular proarrhythmic effects of quinidine were reported as early as in 1922. Systematic studies reveal an incidence of about 5 % [14]. Despite its unfavorable adverse effects, quinidine is still recommended when other drugs have failed to convert AF since there is no doubt about its remarkable efficacy (see Chap. 48).
Quinidine and its congeners were later classified as so-called class I drugs since they slow maximum upstroke velocity of cardiac action potentials, indicating sodium channel block. The obvious efficacy of quinidine has gained enormous interest to search for class I drugs. The result of this effort resulted in a dozen of different class I drugs. From the point of cardiac cellular electrophysiology, they differ with respect to their effect on action potential duration (APD). So class I drugs were later subclassified (e.g., IA, IB, and IC; prolongation, shortening or no effect on APD). See Table 50.1 and also Chaps. 48 and 52.
Table 50.1
Summary of drugs used for AF
Drug | Introduced/approved | Class | INa | IKr | IKur | IK,ACh | AR | MR | Noncardiovascular toxicity | Cardiovascular toxicity |
|---|---|---|---|---|---|---|---|---|---|---|
Quinidine | 1918 | IA | + | + | + | + | α1 | + | Thrombocytopenia, cinchonism, pruritus, rash | QRS prolongation, torsades de pointes |
Disopyramide | 1962 | IA | + | + | + | Anticholinergic effects, blurry vision | Congestive heart failure exacerbation, torsades de pointes | |||
Propafenone | 1976 | IC | + | + | β1/2 | Metallic taste, dizziness blurry vision, bronchospasm | QRS prolongation, congestive heart failure exacerbation | |||
Flecainide | 1975 | IC | + | + | Dizziness, headache, blurry vision | QRS prolongation, excess mortality | ||||
Sotalol | 1992 VT/VF 2000 AF | II/III | + | β1/2 | Bronchospasm | Bradycardia, torsades de pointes | ||||
Ibutilide | 1995 | III | + | None | Torsades de pointes | |||||
Dofetilide | 2000 US only | III | + | None | Torsades de pointes | |||||
Amiodarone | 1967 VT/VF only | III | + | + | α1 | Pulmonary (hypersensitivity pneumonitis, chronic interstitial infiltrates), hepatitis, thyroid (hypothyroidism or hyperthyroidism), photosensitivity, blue-gray skin discoloration with chronic high dose, nausea, ataxia, tremor, alopecia | Sinus bradycardia | |||
Dronedarone | 2009 | III | (+) | + | + | α1 β1/2 | Hepatotoxicity, anorexia, nausea; | Sinus bradycardia | ||
Vernakalant | 2010 Europe only | New | + | + | (+) | (+) | Dysgeusia, sneezing, paresthesia | QT prolongation, hypotension | ||
Ranolazine | 2008 | New | +/+ | + | β1/2 | Dizziness, nausea, headache | QT prolongation |
However, it should be noted that the initial intention to search for congeners of quinidine was driven by the unfavorable selectivity profile regarding extracardiac targets and the slow pharmacokinetics of quinidine. Ajmaline, another plant alkaloid, was a next, less toxic follower of quinidine [15]. It gained wide acceptance in Germany after successful self-experiments by the German cardiologist Kleinsorge, and details were published in 1954 [16].
Lidocaine was initially introduced as a local anesthetic in 1946 and first used as an antiarrhythmic agent in 1950 [17]. Its short half-life makes the drug attractive in emergencies, and the drug was used for many years in the clinical setting of acute myocardial infarction (even as intramuscular injection at that time). Lidocaine use in this indication was stopped because of excess mortality [18]. Lidocaine actions seem to be restricted to ventricles (see Chap. 48). Whereas lidocaine can prolong ERP in ventricular tissue [19], it was found almost ineffective in atrial tissue from the same species [20]. The reason for this finding remains unclear. One concern is that higher concentrations are needed to affect atrial relative to ventricular tissue. However, even almost neurotoxic concentrations of lidocaine are not able to convert AF in humans [21].
Disopyramide is another rather curious example. The drug can effectively stop AF, but its profound negative inotropic effects are troublesome [22]. Ironically, because of this “side effect,” disopyramide gained popularity in treating patients with hypertrophic cardiomyopathy. Disopyramide seems to be safe in patients with hypertrophic cardiomyopathy [23]. From the beginning, disopyramide’s marked antimuscarinic effects had limited patient acceptance [24]. However, this initially unwanted effect regained interest in some forms of vagally induced AF [25]. Disopyramide blocks many different potassium channels including IK,ATP [26]. Relevance of potassium channel blockade to its overall activity is unclear. Disopyramide was popular for conversion of AF in the 1980s but is no longer recommended in the North-American and European guidelines. It remains open to question if disopyramide may offer advantages in vagally induced AF.
Propafenone was tested in 1970s in Germany. It is structurally related to propranolol and blocks β1– and β2-adrenoceptors but not muscarinic receptors [27]. Propafenone is therefore classified as a mixture of a class I drug and a nonselective β-adrenoceptor antagonist. It has gained wider acceptance and is still recommended as a second-line drug to convert AF in people without structural heart disease. Taste complaints represent a strange side effect [28].
Flecainide was the last compound labeled as class I drug and represents a remarkable advance in drug development. Flecainide shows high selectivity for sodium channels, and therefore unwanted effects are mainly restricted to sodium channel block in nerve cells. Such adverse effects occur in about 30 % of patients chronically treated with the drug and illustrate general limitations of sodium channel block [29]. Obviously unwanted effects can be reduced by administration on demand instead of continuous drug administration. The drug was initially approved to treat ventricular tachycardia and later for supraventricular arrhythmias. However, severe proarrhythmia in patients treated with flecainide after myocardial infarction stopped its use in this indication [30]. Putative causes for flecainide’s proarrhythmic action are discussed in hundreds of review articles (including in Chap. 48 of this book); however, the exact mechanism remains unclear. It should be noted that flecainide proarrhythmia could not be reproduced in a sophisticated rat model of myocardial infarction [31]. The latter rather surprising finding led to the idea that the proarrhythmic effect of flecainide may be mediated rather via blockade of other channels than sodium channels such as the hERG (not functional in rat heart). Nevertheless, use of flecainide is no longer accepted in patients with compromised ventricular function. Since many younger patients suffering from AF are free of structural heart disease, flecainide is becoming a popular drug in such patients. There is a consensus that flecainide seems to be safe in this population [32].
50.3.2 Class II Drugs
The β-adrenoceptor antagonists were initially reported as class II antiarrhythmic drugs. Obviously, they do not affect excitability of cardiomyocytes directly, but can prevent harmful arrhythmogenic actions of endogenously released catecholamines. Therefore, they are not able to stop arrhythmias in general. Efficacy of prophylactic β-adrenoceptor antagonist treatment is restricted to situations where AF is the result of acute stress such as postoperative atrial fibrillation [33]. Besides this indication, the efficacy of β-adrenoceptor antagonists in AF is not convincingly proven. Even in the setting of heart failure, where catecholamine levels are clearly elevated, β-adrenoceptor antagonists do not have any impact on the occurrence of AF [34, 35].
50.3.3 Class III Drugs
Sotalol (MJ 1999) is the oldest class III drug [36]. It was initially developed as a competitive β-adrenoceptor antagonist and was intensively tested for its ability to prevent ventricular arrhythmias in the setting of experimental myocardial infarction (so-called class II action) [37]. Somewhat unexpected for a β-adrenoceptor antagonist, sotalol was not only effective against arrhythmias but caused a remarkable positive inotropy. The surprising finding could be later explained by APD prolongation. Sotalol blocks potassium currents that are active during the plateau phase (later identified as IKr, conducted via hERG channels) (see Chap. 48). The use of sotalol was not widely propagated until the early 1990s, when class I drugs had lost acceptance in ventricular arrhythmias and the compound was believed to fill the gap. To avoid the harmful negative inotropy of β-adrenoceptor antagonists that are given acutely in patients with heart failure, the (d)-enantiomer of sotalol, devoid of adrenoceptor antagonist activity but still blocked potassium channels, was introduced to clinical use. However, it became quickly clear that (d-)-sotalol provoked severe ventricular proarrhythmia not only in patients with heart failure [38] but also in an animal model with left ventricular hypertrophy [39]. This quickly resulted in the loss of status for racemic sotalol in the treatment of ventricular tachycardia. Subsequently, sotalol was tested for its anti-AF activity [40]. Racemic sotalol is still frequently used for the prevention of reoccurrence of AF [41]. Its role in converting recent-onset AF is less popular. There are theoretical considerations in the use of sotalol for that application. Any (even electrical) conversion of AF can be followed by severe sinus bradycardia or even sinus arrest [42]. β-Adrenoceptor antagonists are expected to aggravate such a scenario. Since most of the class III agents show the phenomena of reverse-use dependence with excessive APD prolongation at low heart rate, occurrence of torsades de pointes arrhythmia is facilitated [43]. This phenomenon was realized as a dangerous scenario since the 1960s when quinidine (a potent hERG blocker) was frequently used for conversion of AF [44]. From this background, a fixed combination of hERG block with a β-adrenoceptor antagonist may be at least questionable. Nevertheless, application in patients with AF is still popular, for example, in the United States [45, 46]. There is a consensus that sotalol should not be used in case of left ventricular hypertrophy or left ventricular failure.
Amiodarone is another agent classified as a class III antiarrhythmic drug. It was initially developed as a vasorelaxant. The very first study about amiodarone effects on AP showed marked APD prolongation without obvious effects on upstroke velocity [47]. Therefore, amiodarone was classified as a so-called class III drug (APD prolongation only). More than 15 years later, patch-clamp studies revealed sodium as well as calcium channels blocked by amiodarone [48]. Sodium channel block was later found to be use dependent and stronger at depolarized membrane potential [49, 50]. Thus, amiodarone shares typical class I drug features. In addition to the block of ion channels, amiodarone acts as an antagonist at β-adrenoceptors [51]. The relative contribution of different targets affected for the overall activities is less clear, but the blockade of sodium channels may play an important role (compare dronedarone section). Severe hypotension that results because of vasorelaxation, might complicate its use, when given as rapid intravenous injection. The biggest advantage of amiodarone compared to class I drugs is its low incidence for ventricular proarrhythmia [52]. The reason for the difference compared to other class III drugs is unclear [43]. The almost unique feature among antiarrhythmic drugs made amiodarone very attractive when it becomes clear that class I drugs have failed in patients with ventricular arrhythmias and structural heart disease. Consequently, amiodarone was approved for the treatment of life-threatening ventricular arrhythmias. After successful introduction of implantable cardioverter/defibrillators, amiodarone (like any other antiarrhythmic drugs) has lost popularity in this field (Chap. 8). Amiodarone not only suppresses ventricular arrhythmias but also prevents the recurrence of AF. In addition, amiodarone can convert AF; however, the onset of effect is delayed compared to class I drugs (hours vs. minutes). Low propensity for ventricular proarrhythmia makes amiodarone very attractive for treating AF, but the drug was never approved for this indication. Nevertheless, off-label use to prevent reoccurrence of AF is enormously popular (~60 % of all prescriptions in one US population) [45], but needs careful consideration given its extracardiac toxicity. Some of the unwanted effects are rather irreversible and even life threatening. Many of the amiodarone toxic effects are described as cumulative toxicity, and that could be reduced by lower dosages and shorter duration of treatment. Obviously, amiodarone use in younger patients is problematic. Large retrospective trials suggest that a remarkable number of younger patients are treated with amiodarone. Many of them are expected to be free of structural heart disease and could therefore be treated with less toxic class I drugs [46]. There are other issues that need to be addressed. In case of AF, relapse under continued treatment with amiodarone, the indication to continue drug application has to be carefully checked. There is no doubt that amiodarone can slow AV conduction and thereby control rate during AF. However, drugs much less toxic can achieve this goal; amiodarone is not recommended for this indication. Besides, in any controversy about the appropriate indication for amiodarone in AF, there is a general consensus that chronic amiodarone treatment needs intense follow-up. Patients have to undergo several clinical and biochemical tests at a fixed time schedule [53]. One of the most disastrous side effects of amiodarone is its thyrotoxicity. Therefore, it is logical to search for a compound that is structurally related to amiodarone but free of iodine.
Dronedarone is the result of such effort, and like its congener amiodarone, dronedarone blocks a number of different ion channels and adrenoceptors. From the viewpoint of cellular electrophysiology, dronedarone shares many acute effects of amiodarone (e.g., APD prolongation), but it lacks pronounced sodium channel block as seen after chronic treatment with amiodarone [54]. Based on clinical results with amiodarone, it is logical to use dronedarone in patients with heart failure in order to reduce sudden cardiac death. However, the drug failed in this indication because of causing excess mortality due to worsening of heart failure [55], and it is noteworthy that the dronedarone, in contrast to its closely related parent compound amiodarone, was never approved for ventricular arrhythmias. The next step was testing dronedarone for its activity in AF. Like amiodarone, dronedarone is able to convert recent AF [56], but dronedarone, too, was never approved for that indication. Dronedarone prolongs the time to AF recurrence after conversion of AF [57], and the drug was approved by fast track in 2010 for that indication. Dronedarone is clearly less effective compared with amiodarone [58]. In rare cases, dronedarone can induce liver injury, including even several cases of acute liver failure leading to liver transplants [59]. Recently FDA addressed these life-threatening events in a warning letter (http://www.fda.gov/Drugs/DrugSafety/ucm240011.htm). Dronedarone can increase mortality in some patients with permanent AF, and serious safety concerns lead to restrictions in its use (http://www.fda.gov/Drugs/DrugSafety/ucm264059.htm). Dronedarone is recommended as first-line therapy to prevent reoccurrence of AF. However, even if excess mortality is restricted to some patient populations (severe heart failure, persistent AF), it remains questionable whether a drug that can cause such harm should be prescribed for an arrhythmia that is not directly life threatening. Dronedarone has many pleiotropic effects, which are recently summarized in an excellent review [60]. What role of dronedarone should be in the treatment of AF remains to be determined. However, one can conclude that the initial hope to get a substitute for amiodarone that combines the advantages of amiodarone (efficacy against VF and AF without ventricular proarrhythmic potency) with the advantages of advanced drug design (freedom of extracardiac toxicity) could not yet be achieved.
Dofetilide and ibutilide are structurally related to the experimental hERG blocker E-4031, and all of these compounds show high selectivity. In contrast to class I drugs, pure hERG blockers do not lead to CNS side effects. Both dofetilide and ibutilide can effectively stop AF and prevent reoccurrence of AF [61, 62]. However, since blockade of hERG channels is now recognized as the leading cause for drug-induced proarrhythmias in general [63], the acceptance for that drug class is very limited (see Chaps. 48 and 49). In some countries, both drugs were never approved for AF. On the other hand, several studies report dofetilide as a safe drug as long as dosage is adjusted carefully based on QT measurements even in patients with left ventricular dysfunction [64].
50.4 Novel Targets Sites: Atrial-Selective Ion Channel Block
50.4.1 Potassium Currents
50.4.1.1 Ultrarapidly Activating Potassium Current (IKur)
In the human heart, the shape of the action potentials differs remarkably between atrium and ventricle (Chap. 46). While ventricular action potentials show a long-lasting plateau phase at positive membrane potentials, atrial repolarization is characterized by a pronounced phase I repolarization leading to a deep notch with a plateau voltage clearly in the negative range. One reason for this difference is the existence of IKur, encoded by Kv1.5, in atria only [65]. Therefore, blockade of IKur represents a promising example of an atrial-selective ion channel block target [66]. IKur blockade increased the force of contraction in atrial tissue from patients in SR as well as in AF [67], and this helped to reverse contractile dysfunction in AF. However, there are several limitations. Whereas the first report in a goat model of AF showed a clear increase in refractoriness upon mixed Ito/IKur block by AVE0118 [68], the same compound rather shortened APD90 in atrial tissue obtained from patients with SR [69]. Reduced expression of Kv1.5 in AF and inactivation of IKur at higher rates in AF [70] have questioned the value of Kv1.5 as a drug target for AF. Besides this theoretical consideration, Kv1.5 blocker prolongs APD90 in AF. Recent in vitro study implicate effectiveness in human atrial tissue even at higher rates underlying the importance of rather complex interactions complicating prediction of drug effects based on patch-clamp data. One of the most advanced IKur blockers such as D0103 (Xention Ltd, UK) has now progressed to first studies in man [71].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


