Traditional Diuretiocs and Other Diuresing Agents
Stephen S. Gottlieb
Diuretic meications are deemed essential for the treatment of most patients with heart failure, particularly when congestion is present. The underlying abnormality present in patients with heart failure leads to neurohormonal activation as well as salt and water retention. Thus, medicines that increase sodium and water excretion can be extremely effective. Indeed, symptoms of congestive heart failure may be completely relieved by this simple and inexpensive intervention. However, the adverse consequences associated with the use of diuretic medications are rarely considered. Knowledge of potential problems as well as benefits can lead to safer and more effective use of these agents.
Effects of Diuretics on Cardiac Performance
Diuretics have the ability to decrease many of the symptoms associated with heart failure. The presence of systemic congestion leads to abdominal fullness, bloating, and peripheral edema. Whereas the former leads to gastrointestinal (Gl) symptoms (and may impair the absorption of nutrients and medications), the latter is often painful and can greatly limit mobility. Pulmonary congestions leads to the subjective complaint of dyspnea (starting with exertion and progressing to rest) and, when advanced, can impair oxygenation. Diuretics can dramatically improve these symptoms by enhancing the urinary excretion of salt and water. Although this may occur without increasing cardiac index (1), diuretic medications also have been reported to improve cardiac performance at rest and during exercise (2). No direct inotropic effect has been ascribed to any diuretic but there are multiple explanations as to why diuresis may acutely increase cardiac performance.
It is possible that some of the benefits seen with diuretics are secondary to a reduction in systemic vascular resistance. Diuretic-induced reductions in extravascular pressure may directly lead to vasodilation. Improved heart failure status could also lead to a reduction in neurohormonal activation and a withdrawal of the vasoconstriction that is mediated by most of the neurohormonal agents that are increased in heart failure. In addition, venous capacitance increases within 5 minutes of furosemide administration, suggesting an acute mechanism of vasodilation as well (3). In contrast to the decreased peripheral resistance frequently observed following diuresis, however, some of the neurohormonal changes associated with diuretics may cause vasoconstriction. Thus, blood pressure may acutely increase with diuretics, with the beneficial hemodynamic effects of diuresis only developing later (4). Nevertheless, in most patients chronic diuretic use causes decreased symptoms of heart failure, improved cardiac performance, and vasodilation (5). The decreased afterload probably explains many of the reported improvements in cardiac index and other load-dependent indexes of cardiac performance following diuresis (6). The importance of afterload reduction as the cause of the improved cardiac performance is supported by the finding that the increased stroke volume often seen with diuretics is closely related to decreases in systemic vascular resistance, but not to decreases in systemic vascular resistance, but not to decreases in preload (7).
Despite the importance of afterload reduction, there are some actions on preload that also could improve cardiac performance. The abnormal valvular and papillary muscle geometry caused by ventricular enlargement can lead to abnormal mitral valve function and significant
regurgifation (8). Even abnormal atrial geometry may lead to mitral regurgitation (9). Decreases in ventricular (and perhaps atrial) size could therefore increase forward flow by reducing backward flow. The importance of reversing mitral regurgitation has been clearly demonstrated using vasodilators (10). Presumably, diuretic-induced volume reduction also decreases mitral regurgitation and thereby increases cardiac output. This concept could explain why diuretics may improve cardiac output in patients with congestive heart failure but not in individuals with normal cardiac size and function (11).
regurgifation (8). Even abnormal atrial geometry may lead to mitral regurgitation (9). Decreases in ventricular (and perhaps atrial) size could therefore increase forward flow by reducing backward flow. The importance of reversing mitral regurgitation has been clearly demonstrated using vasodilators (10). Presumably, diuretic-induced volume reduction also decreases mitral regurgitation and thereby increases cardiac output. This concept could explain why diuretics may improve cardiac output in patients with congestive heart failure but not in individuals with normal cardiac size and function (11).
Although frequently hypothesized, it is unlikely that a decreased preload shifts the ventricle to a more optimal position on Starling’s curve. A descending limb of the curve has only been noted at extremely high pressures (above 60 mm Hg) (12), and sarcomere length in a dilated canine heart is close to optimal (13). However, it is likely that decreased systolic wall stress with reduced left ventricular size (the Laplace effect) is beneficial. Not only would a decreased afterload improve cardiac performance but the lower filling pressures associated with diuretics might limit ischemia (14). Neurohormonal activation and a consequent positive inotropic effect could be another possible explanation of the improved cardiac performance frequently noted with the administration of diuretics to patients with heart failure.
Diuretics, of course, can lead to decreased cardiac output if the left ventricular filling pressure is decreased excessively. In contrast to the upper end of Starling’s curve, the lower end of the curve is clinically important and it has been demonstrated to be operative in humans (15). Although neurohormonal activation may minimize the effects of the Starling phenomenon in normal individuals, in the severely compromised heart failure patient overdi-uresis may cause devastating effects and should be avoided.
The chronic effects of diuretics on cardiac function may be different than the acute effect. Indeed, there is evidence that chronic furosemide may lead to decreased cardiac function. In a study of rats with pacing-induced cardiomy-opathy, those animals randomized to receive furosemide experienced more cardiac dilatation and decreased contractility than those who received placebo (16). These animals also demonstrated activation of the renin-angiotensin-aldosterone axis (Fig 24-1).
Neurohormonal Effects
Neurohormonal activation is important to the pathophysi-ology of congestive heart failure. It may reflect the severity of disease as well as alter its natural history. These issues are discussed in other chapters but are important to any discussion of diuretics because neurohormones may be affected by their administration. Whether these neurohormonal changes directly cause benefit or harm, or whether they merely reflect changes in physiology, is often debated.
Renin-Angiotensin-Aldosterone System
Stimulation of the renin-angiotensin-aldosterone system (RAAS) is commonly found in patients with congestive heart failure, is associated with increased mortality, and may have both beneficial and detrimental effects. Although stimulation of this system is usually assumed to be secondary to the severity of illness, increased angiotensin II concentrations and plasma renin activity actually often reflect the intensity of diuretic treatment. Renin release may be caused by volume contraction and stimulation of baroreceptors and the macula densa (17). Thus, it is not surprising that many studies document increased plasma renin activity and plasma concentrations of angiotensin II in heart failure patients and animals following diuretic therapy (18,19) (Fig. 24-1). Indeed, concentrations may be normal in heart failure patients not treated with diuretics (18,20).
Aldosterone concentrations are similarly affected by diuretics. Normal concentrations prior to treatment may be
followed by increases with diuresis, (20,21). Interestingly, patients with elevated aldosterone concentrations prior to treatment with diuretics may exhibit decreases following their administration (18). The varying response of aidosterone may reflect the conflicting consequences of the effects of diuretics on the severity of illness and their effects on delivery of sodium to the distal tubule. The importance of both of these factors is suggested by a report that patients with elevated baseline aidosterone concentrations demonstrate decreased concentrations with initial diuretic treatment (and improvement in symptoms), but increased concentrations as dry weight approaches and less sodium is delivered to the renal tubule (22).
followed by increases with diuresis, (20,21). Interestingly, patients with elevated aldosterone concentrations prior to treatment with diuretics may exhibit decreases following their administration (18). The varying response of aidosterone may reflect the conflicting consequences of the effects of diuretics on the severity of illness and their effects on delivery of sodium to the distal tubule. The importance of both of these factors is suggested by a report that patients with elevated baseline aidosterone concentrations demonstrate decreased concentrations with initial diuretic treatment (and improvement in symptoms), but increased concentrations as dry weight approaches and less sodium is delivered to the renal tubule (22).
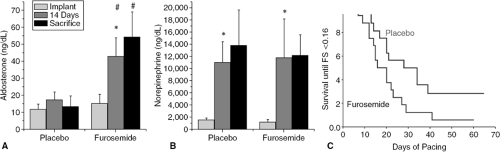 Figure 24-1 Neurohormonal concentrations before and after pacing induced cardiomyopathy in animals receiving placebo or furosemide (Panels A and B). Note that norepinephrine concentrations increased in both groups but aldosterone only increased in those animals receiving furosemide. The animals receiving furosemide also experienced more rapid deterioration in cardiac function (Panel C). (Adapted from McCurley JM, Hanlon SU, Wei SK, et al. Furosemide and the progression of left ventricular dysfunction in experimental heart failure. J Am Coll Cardiol. 2004;44:1301–1307 .) |
The increased angiotensin II and aidosterone caused by diuretic administration may have adverse effects. for example, aidosterone stimulates collagen production (23) and aidosterone antagonists probably have an impact unrelated to actions on electrolytes or diuresis. Indeed, it is likely that the impact of furosemide on the RAAS may explain the decreased contractility caused by furosemide in the rat pacing cardiomyopathy study previously mentioned (16). The implications of the effects of diuretics on the renin-angiotensin-aldosterone axis are presumably mitigated by our interventions with angiotensin-convert-ing enzyme (ACE) inhibitors and spironolactone. Nevertheless, it is possible that RAAS stimulation, even in the presence of antagonists, can cause adverse effects.
Arginine Vasopressin
Following diuretic administration, both stimulation and inhibition of the release of arginine vasopressin (ADH) have been reported. The complex regulation of ADH is probably responsible for these conflicting studies. Arginine vasopressin concentrations are affected by both osmotic and baroreceptor stimuli, and diuretics could increase the concentrations because of actions on these parameters. However, the physiological effects of diuretics are not extensive enough to cause a consistent increase in ADH. Although the decreased blood volume and increased plasma osmolality noted with furosemide (24) might be expected to result in increased arginine vasopressin concentrations, a decrease in volume of 10% is needed to stimulate the baroreceptors and the relative iso-osmotic diuresis observed following the use of most diuretics should not alter ADH concentrations. It is therefore unclear whether diuretics directly cause the increased ADH concentrations often found in individuals with left ventricular dysfunction.
Indirect effects of diuretics, however, could impact on arginine vasopressin regulation; changes in the activity of other neurohormonal systems may alter ADH concentrations. For example, angiotensin II may directly affect its release; this mechanism may explain the increased ADH concentration noted with acute diuresis, even if the diuresis is iso-osmotic and there is no change in oncotic pressure (4). In contrast, normalization of the hemodynamic status with diuresis may return arginine vasopressin concentrations to the normal range (18). Most likely, alterations in arginine vasopressin secretion are not important consequences of diuretic administration.
Atrial Natriuretic Peptide and Brain Natriuretic Peptide
An individual’s volume status is related to secretion of atrial natriuretic peptide (ANP) and brain natriuretic peptide (ANP) and brain natriuretic peptide (BNP). Atrial stretch and ventricular distension cause release of these peptides which, in turn, may cause natriuresis and vasodilation (25,26). Diuretics would therefore be expected to lead to lower concentrations and to mitigation of the peptide’s diuretic and vasodila-tory effects. However, decreased natriuretic peptide concentrations have not been noted following diuretic administration (19) and the high concentrations seen in patients with severe heart failure appear to have little clinical effect, suggesting that downregulation of receptor sites may prevent most of the response to natriuretic peptides in these patients (27). While highly prognostic (28), hemodynamic tolerance to chronic marked elevations in concentration has suggested that any impact of diuresis on natriuretic peptide secretion may be clinically unimportant in these individuals (29). Indeed, the recent ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) trial showed a poor relationship between the change in BNP and the change in pulmonary capillary wedge pressure.
Prostaglandins
Prostaglandins affect the vasculature, and diuretic medications may alter prostaglandin concentrations by both direct and indirect means. Therefore, diuretics may exert beneficial or detrimental actions via this little-appreciated mechanism. For example, furosemide, which increases venous capacitance, does not have the same effect in the presence of indomethacin concentrations known to inhibit prostaglandin production (30). This suggests that its direct actions on prostaglandin production, such as stimulation of the renal production of prostaglandin E2 (PGE2), may have important therapeutic effects. Indeed, the clinical importance of the effects of furosemide on prostaglandin synthesis has been noted in disorders other than congestive heart failure. In premature infants, for example, furosemide increases the incidence of patent ductus arte-riosus, presumably via prostaglandin stimulation (31). Other diuretics also may cause alterations in prostaglandin production; various diuretics increase the production of prostacyclin (PGI2) from arachidonic acid in aortic smooth muscle cells (32). Although the actions of diuretics on prostaglandins is rarely recognized, it is conceivable that some of the beneficial effects observed with diuretics may result from increased concentrations of prostaglandins.
The use of nonsteroidal anti-inflammatory drugs (NSAIDs) may therefore affect the actions of diuretics, especially regarding renal function. Renal prostaglandins help to maintain renal blood flow in the presence of countervailing influences. Thus, if diuresis leads to neurohormonal activation, limiting renal plasma flow, prostaglandins may be essential for the maintenance of renal function. The combination of diuretics and NSAIDs
would then decrease renal plasma flow and glomerular filtration. Indeed, in normal individuals in whom diuretics and aspirin individually had no effect on renal function, the combination caused marked decreases in the glomeru-lar filtration rate (33). In patients with congestive heart failure and compromised renal plasma flow, the effects might be even greater.
would then decrease renal plasma flow and glomerular filtration. Indeed, in normal individuals in whom diuretics and aspirin individually had no effect on renal function, the combination caused marked decreases in the glomeru-lar filtration rate (33). In patients with congestive heart failure and compromised renal plasma flow, the effects might be even greater.
Catecholamines
In contrast to investigations of the renin-angiotensin system, studies of the sympathetic nervous system have consistently reported increased norepinephrine concentrations in patients with heart failure prior to treatment with diuretics (19,20). Also in contrast to the renin-angiotensin system, catecholamine concentrations frequently decrease coincident with the symptomatic improvement caused by diuresis (Fig. 24-1) (20); norepinephrine concentrations are a sensitive reflector of poor cardiac performance (34). This decrease in catecholamines could be an extremely important benefit of diuretic therapy. If catecholamines are responsible for arrhythmogenesis or cardiac deterioration, as is commonly believed (35), diuretics (or any intervention that improves left ventricular performance) may lead to improved myocardial function and survival.
Diuretic-Induced Electrolyte Abnormalities
In addition to the neurohormonal effects, diuretics also have important metabolic actions. Volume contraction leads to avid sodium reabsorption and (when secondary to diuretics other than carbonic anbydrase inhibitors and potassium-sparing agents) bicarbonate reabsorption and metabolic alkalosis. Uric acid excretion is inhibited by the commonly used diuretic medications, and this explains the high prevalence of gout in patients with congestive heart failure. The most problematic consequences of diuretic usage, however, are marked and potentially serious direct and indirect effects on electrolytes. Diuretic medications disturb electrolyte homeostasis, most notably altering potassium, magnesium, sodium, and calcium concentrations. The implications of most of these alterations remain controversial.
Potassium
Most diuretics (other than potassium-sparing agents) increase sodium delivery to the distal renal tubule, where potassium is excreted as sodium is reabsorbed; potassium depletion may therefore result. Although even acute diuresis may cause hypokalemia, total body potassium depletion is associated with the duration of diuretic use (36). Yet, the hypokalemia commonly observed in patients with heart failure cannot be attributed to diuretics alone because hypokalemia is more frequently noted in patients receiving these medications for heart failure than in those receiving them for hypertension. Metabolic alkalosis resulting from intensive diuresis, decreased sodium intake, neurohormonal activation may contribute to the hypokalemic effects of diuretics in patients with heart failure. Activation of the sympathetic nervous system and elevated aldosterone concentrations are also common in patients with severe heart failure and can lead to hypokalemia. Importantly, these factors may stimulate the kaliuretic actions of diuretics even when they do not directly increase potassium excretion. For example, diuretics may potentiate the hypokalemia caused by epinephrine or albuterol (37,38). Thus, although diuretics themselves do not necessarily cause marked hypokalemia, in combination with the abnormal physiology of patients with congestive heart failure most diuretics may induce potassium depletion.
Hypokalemia can cause symptoms such as muscle aches, but of most concern are the potential arrhythmo-genic effects of this state. In the hypertension literature the effect of hypokalemia on mortality has been controversial (39,40) but it is generally accepted that hypokalemia increases the risk of arrhythmias in patients with heart failure. Most investigators assume that studies of hypertensive patients are not relevant to patients with congestive heart failure because of marked differences in anatomy and physiology. Although the concern about the dangers of hypokalemia in patients with heart failure is based on indirect evidence, it is convincing. For example, hypokalemia increases the frequency of early after-depolarizations caused by quinidine (41) and the risk of arrhythmias associated with digoxin (42). Low serum potassium concentrations are also associated with increased risk of arrhythmias following a myocardial infarction (43). More important than the degree of hypokalemia, however, is probably the rate of change of the serum potassium (44); hypokalemia associated with rapid diuresis may be more detrimental than that associated with chronic use of diuretics.
When discussing diuretics, the risks of hyperkalemia are frequently neglected. Not only may the use of potassium-sparing agents lead to hyperkalemia, but the routine prescription of potassium supplements with initiation of diuretics also can cause this fatal complication. Poor renal function, concomitant use of angiotensin-converting enzyme (ACE) inhibitors and/or aldosterone antagonists, and excessive potassium in take can produce hyperkalemia and consequent ventricular arrhythmias and sudden death (45). Therefore, the serum potassium concentration should be monitored carefully in all patients receiving diuretics.
Considering the problems associated with abnormal potassium concentrations, it is wise to keep the serum concentration above 4.0 mEq/L, but within the normal range. Although potassium-sparing diuretics or ACE inhibitors may be used to help achieve this goal, it should be realized that the combination of these two interventions increases the risk of hyperkalemia. Salt substitutes contain potassium and also should be considered as a cause of hyperkalemia in patients receiving potassium-sparing agents. The use of any diuretic mandates close follow-up of the patient in order to maintain the potassium concentration within the normal range.
Magnesium
Although reports of the prevalence of magnesium deficiency in patients with congestive heart failure range from 7% to more than 37%, the true prevalence is disputed for many reasons (46). First there is no gold standard measurement of magnesium deficiency. Determination of the serum magnesium concentration is easy but it may not reflect total body stores of the electrolyte. Serum, myocar-dial, lymphocyte, and muscle magnesium concentrations do not correlate (47) and the concentration (if any) that is most clinically relevant is unknown. There are other factors that also prevent determination of the true prevalence of magnesium deficiency. Diverse definitions of normal magnesium concentrations, differing nutritional intake, and analyses of patients with varying severity of disease have led to wide-ranging estimates of the prevalence of magnesium depletion. Nevertheless, investigations consistently demonstrate that a large percentage of patients with heart failure are magnesium-deficient.
It is often assumed that diuretics are the cause of magnesium depletion in patients with heart failure, and chronic diuretic use probably does lead to magnesium depletion (48), with the duration of therapy an important determinant of the extent of depletion (36). However, other factors also affect magnesium excretion. Some studies suggest that low magnesium concentrations are common in heart failure patients even prior to treatment with diuretics (49), possibly caused by aldosterone-induced magnesium excretion (50). In contrast, other neurohor-monal mechanisms may limit the excretion of magnesium caused by diuretics; renal denervation leads to increased magnesium excretion following the administration of diuretics (51). Despite these confounding factors, it is likely that diuretics are the predominant cause of hypo-magnesemia in congestive heart failure patients.
The clinical implications of magnesium depletion in patients with congestive heart failure are controversial. There are reports of various symptoms related to hypomag-nesemia, ranging from muscle weakness to depression (52). It is also possible that magnesium depletion impairs cardiac performance, either by directly reducing cardiac contractility (53) or by causing peripheral vasoconstriction (54). However, the consequence of most concern is the possibility that magnesium depletion causes ventricular arrhythmias and sudden death.
There are reasons to suspect that magnesium depletion increases the frequency of ventricular arrhythmias. A low magnesium concentration decreases the threshold needed to induce ventricular tachycardia and ventricular fibrillation in normal and digitalis-treated dogs (55), and hypomagnesemia potentiates the development of digitalis-induced arrhythmias (56). Furthermore, administration of intravenous magnesium suppresses early after-depolarizations and ventricular tachycardia induced by cesium in dogs (57) and may prevent the clinical occurrence of ventricular tachycardia in patients (58,59). These actions of magnesium may be related to its function as a critical cofactor in the cellular functions that regulate intracellular electrolyte concentrations (60). Hypomagnesemia also may exacerbate the development of hypokalemia and cause arrhythmias indirectly (61).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


