Fig. 8.1
Risk of AMI associated with number smoked, by age group. P for interaction <0.0001. Nev never smokers, Form former smokers. 1–19 and >20 refer to the number of cigarettes smoked daily (Reprinted from Teo et al. (2006), with permission from Elsevier, Licence no 2905480461542)
Furthermore, smokers who suffer MI are significantly younger than nonsmoking patients with the same condition. A retrospective analysis found that age of onset of first MI was directly related to the amount smoked, with heavy smokers suffering their first MI on average 8.7 years earlier than nonsmokers (Gottlieb et al. 1994). This effect has been suggested to be greater in women than men (Grundtvig et al. 2009). Figure 8.1 shows that while smoking intensity increases the risk of MI significantly, this effect is strongest at a younger age, for example, smoking more than 20 cigarettes per day is associated with a sevenfold increase in risk at age <40, while at age >70 the associated risk is approximately twofold (Teo et al. 2006).
Smoking not only increases the risk of developing CHD but also accelerates its development. Smokers who have already developed CHD have greater odds of mortality than their nonsmoking counterparts, for example, Grundtvig and colleagues (2011) reported 30 % greater post-MI mortality in smokers compared to nonsmokers. In a cohort of 1,521 first MI patients followed up for 13 years, persistent smokers had twice the odds of dying compared to both never smokers and those who had quit prior to MI, after controlling for clinical and socioeconomic characteristics and traditional risk factors (Gerber et al. 2009). Those who quit after MI also earned a survival benefit, reducing their risk substantially.
8.2.1.2 Stroke
Tobacco smoke is also an important cause of stroke. The Framingham study followed a cohort of over 4,000 people for 26 years and reported a significant increase in stroke risk in smokers compared to nonsmokers (Wolf et al. 1988). Additionally, a dose–response curve was evident, with heavy smokers at much greater risk compared to light smokers. Excess stroke risk reduced to nonsmoker levels 5 years after quitting smoking. A meta-analysis of 32 studies investigating the risk of stroke associated with cigarette smoke reported a relative risk of 1.5 (Shinton and Beevers 1989). Stroke risk in smokers is greater during middle age, with a risk of 2.9 under 55 years compared to 1.1 for smokers over 75, when stroke incidence is much higher in the general population. A recent review of studies conducted around the world put the excess risk of stroke in smokers at two to four times that of nonsmokers or those who had quit 10 years previously (Shah and Cole 2010).
8.2.1.3 Sudden Cardiac Death
The Framingham Heart Study reported an increase in sudden cardiac death for smokers compared to nonsmokers with a relative risk of 2.5 over 26 years of follow-up (Kannel et al. 1975). Other studies reported similar associations, for example, an odds ratio of 1.8 for sudden cardiac death in current smokers in the National Mortality Followback Survey (Escobedo and Caspersen 1997). Sudden cardiac death may or may not be preceded by diagnosis of CHD, and it may be the first sign of underlying heart disease. Two-thirds of sudden cardiac deaths due to acute coronary thrombosis occur in cigarette smokers.
8.2.1.4 Dose–Response
It is generally accepted that a dose–response relationship exists between tobacco smoke and adverse cardiovascular outcomes (Fig. 8.2). Indeed, this is one of the features that allowed demonstration of a causal relationship. However, some studies do not support a linear relationship, and there is some evidence of a large effect from a small exposure. A meta-analysis of epidemiological studies found the risk of ischemic heart disease for smoking one cigarette per day to be approximately half that associated with a 20-a-day habit, but still 30 % more than unexposed nonsmokers (Law et al. 1997). Environmental exposure of individuals living with a smoker was similarly associated with a 30 % increased risk of heart disease, although the amount of smoke ingested is around 100 times less than that of an active smoker. This scale of effect from secondhand exposure was calculated as increasing the risk of CHD death in British nonsmoking men aged 60–69 from 5 to 6 % (Law et al. 1997). A prospective study conducted in Norway assessed the risks associated with smoking one to four cigarettes per day and reported that compared to never smokers, light smokers had a surprisingly high relative risk of 2.74 of ischemic heart disease mortality (Bjartveit and Tverdal 2005).
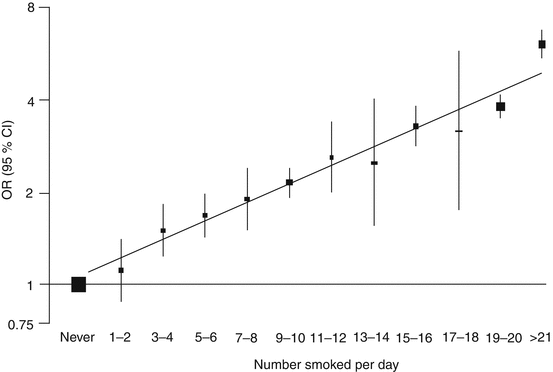
Fig. 8.2
Risk of AMI with increasing number of cigarettes smoked compared with never smokers (Reprinted from Teo et al. (2006), with permission from Elsevier, Licence no 2905480461542)
8.2.1.5 The Illusion of “Light” Cigarettes
In recent years, and due to the indisputable evidence concerning the adverse health effects of cigarettes, low tar or “light” brands have been increasingly marketed as less harmful than ordinary cigarettes. There is some debate however as to the true nature of this harm reduction strategy. A large prospective study found modest reductions in coronary heart disease mortality associated with a 15 mg reduction in tar (Tang et al. 1995). However, a review of research undertaken in this area concluded that only a minority of studies examining cardiovascular endpoints had evidence to support reduced risk associated with low tar cigarettes and that the reduction was at best modest, cited as approximately 10 % (Kabat 2003). Furthermore, in a case–control study of MI patients, no reduction in MI risk was seen in smokers of low-yield cigarettes (Negri et al. 1993). While reductions in cancer risk may be greater due to reduced tar content, cigarette components other than tar, such as nicotine and carbon monoxide, are largely responsible for the increased CV risk seen in smokers. It has additionally been proposed that laboratory smoking machines which test tar yields do not accurately portray the way that cigarettes are smoked and that smokers may compensate when smoking “light” cigarettes by smoking more intensively.
8.2.2 Effects of Secondhand Exposure to Tobacco Smoke on CVD
Secondhand exposure to tobacco smoke, or passive smoking, is known to have severe adverse health outcomes, via the same mechanisms as active smoking. A study of the burden of disease resulting from secondhand smoke in 192 countries estimated that 40 % of children and 34 % of adult nonsmokers are exposed to tobacco smoke worldwide, causing an estimated 379,000 annual deaths from ischemic heart disease (Oberg et al. 2011). Furthermore, almost three million disability-adjusted life years were attributed to CHD caused by passive smoking. Passive smoking is estimated to increase the risk of CHD by approximately 25–30 % (Barnoya and Glantz 2005; He et al. 1999), while the increased risk of an acute cardiovascular event associated with regular exposure to secondhand smoke (SHS) has been reported to be as high as 90 % (Panagiotakos et al. 2002). Even occasional exposure was associated with a 26 % increased risk in the same study. Indeed, acute effects of SHS have been demonstrated within hours of exposure including endothelial dysfunction, reduced heart rate variability, and increased arrhythmia (Kato et al. 2006; Chen et al. 2008). Further studies have shown impaired endothelial function and oxidative stress resulting from exposure to as little as 30 min of SHS (Kato et al. 2006; Raupach et al. 2006; Otsuka et al. 2001; Heiss et al. 2008). It has been estimated that passive smoking causes between 21,000 and 75,000 CHD deaths and up to 128,000 MIs per year in the USA alone, entailing a yearly health bill of $2–6 billion (Lightwood et al. 2009).
A systematic review of the literature reported that the adverse effects of passive smoking can already be seen in children, with exposed children – measured by serum cotinine – showing unfavorable blood lipid profiles and decreased vascular function, predisposing them to develop CVD (Metsios et al. 2010). Randomized controlled trials specifically found impaired endothelial function and aortic elasticity in children exposed to secondhand smoke (Kallio et al. 2007). Inflammatory markers have also been shown to be increased in passive smokers. A British study found that healthy adults with high levels of cotinine from secondhand exposure had higher risk of all-cause and cardiovascular deaths, with around half of the elevated risk explained by higher levels of C-reactive protein (Hamer et al. 2010); while a parallel study found elevated fibrinogen and homocysteine levels associated with passive exposure (Venn and Britton 2007).
Many studies assessing the risks of passive exposure to tobacco smoke involve nonsmokers who live with smokers and are therefore exposed on a daily basis. Research has shown significant increases in both total and cardiovascular death rates in this population (Svendsen et al. 1987; Helsing et al. 1988; Sandler et al. 1989; Garland et al. 1985). Indeed, a review by Wells suggested that “ischemic heart disease appears to be by far the major mortality risk from passive smoking” (Wells 1994). A review of the literature reported the effects of SHS to be almost as large as that of active smoking (Barnoya and Glantz 2005), somewhat contradicting the dose–response theory. While smokers receive about 100 times more smoke than do those affected by secondhand smoke, the risks do not increase in a comparative fashion, for example, relative risks of developing CHD according to a meta-analysis were 1.8 for smokers compared to 1.3 for nonsmokers who live with smokers (Law et al. 1997). A large-scale study of nonsmokers living with smokers followed up for 12 years found a death rate of 1.2 compared to non-exposed individuals, a rate comparative to that of ex-smokers and light smokers and about half the rate of heavy smokers (Sandler et al. 1989). The risk of cardiovascular death was even higher, reported as 1.3 in exposed men and 1.2 in exposed women (Helsing et al. 1988). Multiple investigations have shown a greater risk of SHS exposure at a younger age (Helsing et al. 1988; Sandler et al. 1989).
There are several limitations inherent to research on SHS. Exposed individuals (i.e., living with a smoker) are frequently compared to unexposed individuals (i.e., those not living with a smoker); however, this dichotomy is rather crude, since people may be exposed to SHS in other places such as their place of work. Later studies used biomarkers such as cotinine (metabolized nicotine in the blood) to assess exposure to smoke, with levels in a multicity study indicating that even the “non-exposed” control group had some level of SHS exposure (Whincup et al. 2004). Exposure to passive smoking was reported to be associated with between 68 and 86 % of the risk of light smoking, defined as smoking less than ten cigarettes per day.
8.2.3 Effects of Thirdhand Exposure to Tobacco Smoke on CVD
Recent evidence has accumulated concerning the dangers of thirdhand or environmental tobacco smoke, residual toxic compounds which remain on surfaces and in dust after a cigarette has been smoked. These are then reemitted into the air and react with other compounds, yielding secondary pollutants (Matt et al. 2011). An early study of this issue found household dust to contain modest amounts of nicotine (Hein et al. 1991). Thirdhand smoke is thought to particularly affect children living with smokers, where high levels of smoke pollution can be found on household materials. In a study of households with infants, levels of environmental smoke were five to seven times higher in smoking compared to nonsmoking households even when smokers actively tried to protect their children by smoking outdoors (Matt et al. 2004). Levels in households where smoking was permitted indoors were substantially higher. The health impact of thirdhand smoke remains controversial and requires further investigation.
8.2.4 Effects of Smokeless Tobacco on CVD
Around the world, tobacco habits vary, and in some countries, smokeless tobacco – which includes wet and dry snuff and chewing tobacco – is very common. Since the adverse effects of smoking have come to light and received so much negative press, tobacco companies are seeking new products to sell, including smokeless tobacco, which is marketed as a healthier option. The worldwide Interheart study examined the risk of MI in different types of tobacco users and found that tobacco chewers had more than double the odds of suffering an MI compared to non-tobacco users (Teo et al. 2006). The Atherosclerosis Risk in Communities study also found increased cardiovascular risk in users of smokeless tobacco (Yatsuya and Folsom 2010). The AHA released a statement in 2010 affirming that while smokeless tobacco is less harmful than cigarettes, long-term use is associated with increased risk of fatal MI and stroke, as well as reducing post-MI survival odds (Piano et al. 2010).
8.3 Changes in Smoking: Cessation and Reduction
8.3.1 Effects of Smoking Cessation on the Individual and the Community
Overwhelming evidence has accumulated demonstrating that quitting smoking significantly reduces risks of both cardiovascular mortality and morbidity (USDHHS 1990). Short-term effects of cessation include reduced blood pressure and heart rate (Minami et al. 1999), while in the long term, former smokers can expect to reduce their risk of developing and dying from CVD.
The good news is that even individuals who have smoked for many years can benefit and add years to their life expectancy by quitting smoking at almost any age (Fig. 8.3). After 1 year of abstinence, ex-smokers reduce their excess risk of CHD mortality by half, and after 15 years, the risk returns to that of never smokers (USDHHS 1990). Similar improvements are seen for stroke risk. The Nurses’ Health Study followed 100,000 women for 20 years and reported that compared to continuous smokers, those who quit smoking reduced their risk of death from CHD and cerebrovascular disease by 60 % (Kenfield et al. 2010). This survival benefit increased with time elapsed from cessation. Furthermore, survival benefit is seen even at older ages and in those with established CVD, with reduced risk of reinfarction and mortality, for example, healthy adults aged 60–64 reduced their mortality risk by 10 % after quitting, and post-MI patients with an average age of 54 who quit smoking after MI reduced their mortality risk by 40 % compared to persistent smokers (Gerber et al. 2009). A meta-analysis of 12 cohort studies investigating smoking cessation after MI reported a mortality odds ratio of 0.54 associated with quitting and a relative risk reduction of between 15 and 61 % for mortality (Wilson et al. 2000).
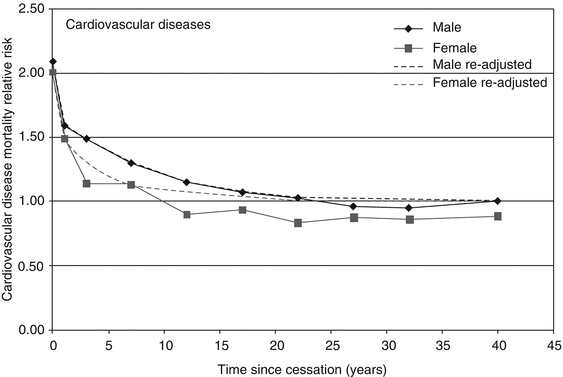
Fig. 8.3
Relative risk of CVD among ex-smokers. Zero years represent current smokers. The estimates at 40 years represent cessation of more than 35 years in the subjects. Data are from ACS CPS-II 1998 follow-up (Reproduced from WHO (2004), permission ID: 101036)
While the health effects of tobacco smoke are largely cumulative, reports show evidence of acute effects too. Reduced admissions for acute coronary events have been consistently reported following implementation of smoking bans (Sargent et al. 2004; Sims et al. 2010; Callinan et al. 2010). In 2011, 55 countries had implemented some kind of smoke-free legislation, as part of the WHO’s tobacco control convention framework. Countrywide or statewide studies have been conducted where smoking bans have been enforced, in order to detect differences in mortality and hospitalizations associated with changes in smoking behavior and exposure. Several meta-analyses have reported significant and seemingly immediate effects of smoking legislation consistently across various countries and cultures, with effect sizes ranging from 10 to 20 % reduction in acute cardiac events. Glantz reviewed eight studies published between 2004 and 2008 in different countries and found a reduction of 19 % in hospital admissions for acute MI associated with smoking legislation (Glantz 2008). Reductions in secondhand smoke exposure resulting from smoking legislation were even higher, averaging between 64 and 84 %. The benefits of smoke-free legislation seem to accrue with time; for instance, after 12 months, the rate of acute cardiac hospitalization had reduced by 15 % and after 3 years by 36 % (Lightwood and Glantz 2009). Another meta-analysis that reviewed 17 studies reported a pooled estimate of a 10 % reduction in acute coronary syndromes following smoking bans, improving by 6 % each year (Mackay et al. 2010). A third review of 11 international studies reported an overall decrease of 17 % in MI risk, emphasizing greater risk in younger people (Meyers et al. 2009). While some problems are inherent in these types of studies, including limited exposure assessment and short-term follow-up, results consistently show reduced cardiac risk following the introduction of legislation protecting people from exposure to tobacco smoke.
8.3.2 Smoking Reduction
Smoking cessation is the best way to reduce cardiovascular risk for smokers. However, since quit rates are low and many smokers are either unable or unwilling to quit, several studies have investigated whether a reduction in smoking intensity is associated with significantly reduced cardiovascular risk. While this issue remains controversial – indeed, the latest US Surgeon General’s report does not advocate smoking reduction for CVD risk reduction (USDHHS 2010) – there is some evidence to support this theory. Several studies have demonstrated short-term benefits of reduction with regard to cardiovascular risk factors such as lipids and blood pressure (Pisinger and Godtfredsen 2007). A reduction of daily cigarette intake in MI survivors was related to decreased mortality risk; each reduction of five cigarettes per day was associated with an 18 % decline in mortality risk (Gerber et al. 2009). Furthermore, in a cohort of working men followed up for 40 years, a reduction in smoking intensity was associated with increased odds of reaching age 80 and a 15 % reduction in mortality, with the largest benefit for cardiovascular survival (Gerber et al. 2012). Despite this evidence, several large-scale studies found no significant survival benefit, or reduced risk of MI, due to smoking reduction (Tverdal and Bjartveit 2006; Godtfredsen et al. 2003). It has been hypothesized that a reduction in the number of cigarettes may not always equate to a reduction in the intake of nicotine or other components, due to compensatory smoking, whereby smokers smoke each cigarette more intensely and for longer (Godtfredsen et al. 2006). This issue requires further investigation before the benefits of smoking reduction can be dismissed.
8.4 Mechanisms
Tobacco smoke has both acute and chronic effects, being associated with vascular inflammation and autonomic dysfunction (Wells 1994). The components of tobacco smoke which increase the risk of developing CVD are oxidizing chemicals, nicotine, carbon monoxide, and particulate matter which together produce a chronic inflammatory state (USDHHS 2010). Short-term effects include decreased platelet sensitivity leading to increased platelet aggregation and decreased oxygen supply to the heart (Wells 1994). Platelet aggregation and blood viscosity affect the blood lipid profile in the long term, increasing triglycerides and lowering high-density lipoprotein cholesterol (Blache et al. 1992), all factors which contribute to the development of CHD. Of the many toxic components of tobacco smoke, nicotine seems to be largely responsible for the change in platelets and increased heart rate and blood pressure (Davis et al. 1985), while the reduction in the blood’s ability to supply oxygen is due to a buildup of carbon monoxide (Wells 1994). Indeed, men who lived with smokers were found to have raised levels of expired carbon monoxide compared to controls (Svendsen et al. 1987). Longer-term effects of smoking begin from damage to the arterial endothelium and involve a buildup of plaque leading to atherosclerosis, increasing the risk of thrombosis, often detected via carotid artery wall thickness, and an imbalance of blood lipids.
The key pathways through which tobacco smoke causes and exacerbates CVD are atherosclerosis and endothelial injury (USDHHS 2004). Atheroma, the underlying pathological process preceding most coronary and cerebrovascular events, develops over years or decades. Atherosclerosis underlies CHD and involves a hardening of the arteries due to fat deposition and thickening of arterial walls, which cause a narrowing of the lumen and consequently reduced blood flow. A thrombus or clot can then form and break off, causing an infarct or stroke. The toxic components of tobacco smoke affect these processes. The 2004 Surgeon General’s report found sufficient evidence to infer a causal relationship between smoking and subclinical atherosclerosis, the early development of atherosclerosis before clinical symptoms manifest (USDHHS 2004). The effects of smoking can be seen in the carotid and popliteal arteries, with greater presence of atherosclerosis. While earlier studies examined the correlation between smoking and cardiac endpoints such as MI or cardiac death, more recent research has investigated earlier effects which begin the cascade of events and go further in explaining the pathogenic pathways. Carotid intima medial thickness is often used as a marker of atherosclerosis since it is a subclinical sign yet is related to both CHD and stroke. Coronary calcium can also be measured to assess the presence of atherosclerosis. The Atherosclerosis Risk in Communities study followed up healthy participants for 3 years and found that smoking was strongly related to carotid atherosclerosis after controlling for age, with smokers showing greater intima medial thickness and incidence of CHD events (Sharrett et al. 1999). Studies have shown that atherosclerosis is already present at a young age, for example, an autopsy study of young people aged 15–34 showed atherosclerotic lesions in the carotid artery of the majority, increasing with age (Strong et al. 1999). Lesions have been shown to progress more rapidly in smokers compared to nonsmokers.
8.4.1 Endothelial Dysfunction
The endothelium, the upper layer of the arterial bed in contact with blood flow, controls vasodilation and constriction. Endothelial damage caused by tobacco smoke can impair the vessels’ ability to dilate, at the same time increasing inflammation and cell proliferation, which contribute to the development and progression of atherosclerosis (Barnoya and Glantz 2005). Evidence from umbilical arteries and from the uterine arteries of pregnant smokers showed changes in the endothelium including intracellular holes and widening of intercellular junctions. Further studies in smokers showed elevated levels of circulating endothelial cells after smoking two cigarettes (Davis et al. 1985). Markers of endothelial damage such as von Willebrand factor have been shown to be increased minutes after smoking (Blann et al. 1998). Oxidizing chemicals and nicotine are thought to be responsible for endothelial dysfunction (USDHHS 2010). Once the endothelium is damaged, platelets rapidly stick to it and to each other, thereby increasing the risk of thrombosis (Meade 1994).
8.4.2 Inflammation
Exposure to tobacco smoke causes elevations in inflammatory markers. Cigarette smoking produces a chronic inflammatory state that contributes to the atherogenic disease processes. C-reactive protein is elevated in smokers, even years after quitting (Tracy et al. 1997). The same inflammatory marker has been show to predict CHD events. Fibrinogen, which is elevated in CVD, is strongly linked to smoking, with smokers displaying much higher levels than nonsmokers (Kannel et al. 1987; Miller et al. 1998). The level of fibrinogen increases with the amount smoked. Fibrinogen levels increase on starting smoking and decrease on cessation but remain somewhat elevated in ex-smokers for 5–10 years (Meade 1994; Rothwell et al. 1991).
8.4.3 Lipids
Widespread evidence, including meta-analyses, has demonstrated that smoking is associated with adverse lipid profiles (Craig et al. 1989), particularly high concentrations of low-density lipoprotein cholesterol (LDL) and reduced high-density lipoprotein cholesterol (HDL). Furthermore, HDL levels – which are essential in preventing atherosclerosis – were shown to decrease after starting smoking and to increase after quitting (Fortmann et al. 1986). Cigarette smoking produces an atherogenic lipid profile, primarily due to an increase in triglycerides and a decrease in HDL (USDHHS 2010). SHS has been shown to have the same effect, causing reduced HDL following both chronic (Mizoue et al. 1999) and acute exposure (Moffatt et al. 2004).
8.4.4 Cortisol/HPA Axis
Tobacco smoke causes an increase in cortisol secretion (Direk et al. 2011) and blunted cortisol response to stress (Rohleder and Kirschbaum 2006). Cortisol, produced by the hypothalamic–pituitary–adrenal (HPA) axis, is related to stress and to numerous chronic illnesses. Inhibition of cortisol response may be responsible for the increase in atherosclerosis seen in smokers. Chronic exposure to nicotine increases the activity of the HPA axis which, being involved in inflammatory pathways, may lead to increased inflammation and thereby contribute to CV morbidity (Rohleder and Kirschbaum 2006).
8.4.5 Blood Pressure
Blood pressure increases almost immediately during smoking, with the elevation largely caused by nicotine (Omvik 1996). Numerous studies have demonstrated short-term effects of tobacco, for example, Barutcu showed increased blood pressure and heart rate after smoking a single cigarette (Barutcu et al. 2004); and a study of the short-term effects of smoking cessation found blood pressure to be decreased during 1 week of abstinence compared to usual smoking behavior (Salonen et al. 1981). While epidemiological studies have not demonstrated chronically elevated blood pressure in smokers or have reported only weak associations (Bowman et al. 2007), results in an animal model demonstrated significant hypertension resulting from prolonged exposure to cigarette smoke (Talukder et al. 2011).
8.4.6 Platelet Aggregation
Studies of environmental exposure to tobacco smoke have shown a relationship with platelet aggregation, seen immediately after exposure and even at low doses (Law et al. 1997). Twenty minutes of exposure to SHS was associated with an increase in platelet aggregation related to a 34 % increased relative risk of CHD. SHS has been shown to activate blood platelets thereby increasing the risk of thrombosis (Barnoya and Glantz 2005). Further evidence of this causal pathway is the increased fibrinogen detected in passive smokers (Iso et al. 1996), since fibrinogen is a mediator of platelet activation, as well as an inflammatory marker.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


