Fig. 2.1
Progressive decrease of overall yearly mortality in successive heart failure (HF) trials, where beta-blockers (beta Bl) [12–14] and, later, cardiac resynchronization therapy (CRT) [15] were tested. Notably, over the course of time, the selected HF populations had progressively more severe disease (this was based on patient selection criteria and confirmed by the worse one-year mortality). The key information stands in the fact that, over time, each treatment was able to step back the patient study outcome to the one-year mortality observed in the control arm included in the precedent study
Similarly, the implantation of a lifesaving implantable cardiac defibrillator (ICD) following the Multicenter Automatic Defibrillator Implantation Trial (MADIT) criteria does not complete the course of HF therapy as many expected. Adequate prevention of sudden death, indeed, dropped overall mortality of 5.6 %, but, after the effective delivery of the defibrillation therapy, the disease paradoxically progresses [14].
After reputed experts trumpeted outstanding success in HF management, the crude data confirm we are just able to curb disease progression in a portion only of the HF population, but we are unable to fully reverse and definitively cure the disease. More dangerously, we are still led by some misleading concept in daily practice. It is the case of how acute HF management is currently widely performed. After the abrupt development of symptoms driven by lung congestion in the vast majority of cases, indeed, diuretic drugs remain the pivotal therapy [15].
Physicians persist in staggering diuretic drug dosing despite they know congestion is the consequence of a number of failures that overrun compensatory mechanisms of the whole cardiovascular setting. Common knowledge addresses that despite dyspnea is driven by lung congestion, it is a flashing signal of the inadequate pump function that critically pounds kidney perfusion. In the effort to provide rapid relief to the patient’s dyspnea, that is poorly treated by rapid diuresis [16], doctors often increase diuretic dose over-sighting that kidney function can deteriorate [17] and that this consequence will decrease drug efficacy [18], worsening patient outcome [19].
On note, the critical balance between the kidney perfusion and the blood pressure becomes a crucial factor in the advanced HF, when even a modest reduction of systolic blood pressure runs disproportionate fall in the renal performance (Fig. 2.2) [20].
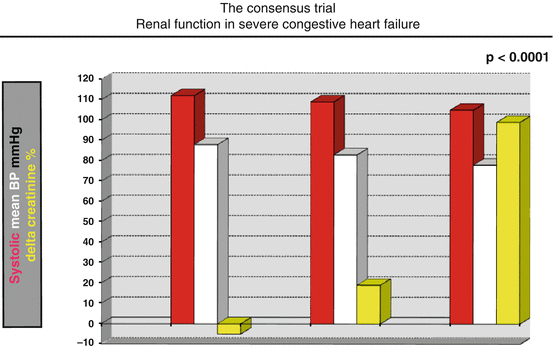
Fig. 2.2
The tight relationship between arterial pressure and renal failure is clearly highlighted by the CONSENSUS study data. When arterial pressure falls below a threshold value, kidney function strikingly worsens. In this figure, the mean arterial pressure fall from 90 to 80 mmHg serum leads to a creatinine increase of 100 % from [20]
These data, collected in an advanced HF population of the CONSENSUS (Cooperative North Scandinavian Enalapril Survival Study) trial, address the relationship between blood pressure and kidney function and may become the critical crossover between therapy benefit and therapy adverse events.
This is because renal dysfunction, per se, plays a direct role in the development and progression of HF and the majority of patients hospitalized for acute decompensated HF have been shown to have already some degree of renal dysfunction [21].
More importantly, renal failure is a more powerful predictor of HF outcome than pump performance indexes like LVEF [22].
Physicians are frequently blurred by patient symptom and they do not mind the underlying pathophysiological key of the disease: the arterial vasculature under-filling. The main consequence of poor cardiac performance, indeed, is the low cardiac output that decreases the kidney perfusion in order to spare the heart and the brain circulation, thereby disproportionately decreasing the renal fraction of cardiac output [23].
One critical consequence of the greater imbalance in renal perfusion is the consequent disproportionate enhancement of renal sympathetic afferent/efferent nerve activity that results in marked increases in renal norepinephrine spillover, with a sympathetically mediated increase in plasma renin activity [9, 23].
In addition to efferent sympathetic activation, activation of renal sensory nerves in HF may cause a reflex increase in sympathetic tone that contributes to the progression of HF by targeting the function of other end-organs, namely, heart and vessels, including venous capacitance in the splanchnic organs [24, 25].
Loop diuretics currently administered in order to clear off fluid volume overload act as a double-edged sword. On one side, they increase water and sodium excretion slowly providing congestion relief [26], but on the other, they promote hypoosmotic diuresis contributing to the water/sodium plasma unbalance [27].
Such an unbalance and fluid loss will eventually have consequences on cardiac output and renal perfusion [17]. This unbalance will indeed create the optimal condition for a vicious circle leading to a further augmentation of the sympathetic/renin-angiotensin system activation with obvious further detrimental consequences on renal perfusion [28] and ultimately in HF outcome. This is the pathophysiology underlying the dramatic negative prognostic consequence of high loop diuretic daily dose [29] and it becomes one more killer in face of the re-uprising of life losses.
More recently, in the effort of improving the patient outcome, several randomized controlled studies have been performed testing plasma concentration of variations of B-type natriuretic peptide (BNP) or of its amino-terminal metabolic product N-terminal-proBNP (NT-proBNP) as a specific guide for up-titration of neurohormonal drugs and optimization of loop diuretics [30].
Only in the ProBNP Outpatient Tailored Chronic HF Therapy (PROTECT trial) NT-proBNP–guided care was associated with a significant reduction in total cardiovascular (CV) events, including worsening heart failure (HF), hospitalization for HF, and CV death. The overall mortality reduction reached almost the statistical significance in patients younger than 75 years but failed to add benefit in the older population [31]. These results should not discourage an appropriate use of biomarkers to optimize lifesaving therapies but do emphasize the need for a better understanding of individual variables that really count in the setting of HF.
In the attempt to turn the tide, today, we can implement sophisticated technologies in treatment of selected cases, such as left ventricular assist devices (LVADs). Those technologies are now a suitable option in experienced HF centers since they displayed impressive implementation involving size, weight, dependability, durability, and implant technique. Skill of surgery team, patient selection criteria, device selection, post-implant patient management, and education are also much improved. Nevertheless bad news are raining again on the end of the story, LVAD chance is most linked to the therapy cost and its burden remains far from a fair cost-effectiveness balance [32].
Moreover, given the need of a major surgical approach and of the complex postoperative management, this sophisticated high-cost “halfway technology” therapy remains, so far, an option only for a tiny minority of patients. The vast majority of those who experience progressive worsening of HF symptoms are old and/or they cluster a number of comorbid conditions (more than three in the average [33] that prevent them to be the ideal LVAD candidates). In the largest advanced HF population, the current prospective remains bleak. The costs due to increased physician visits, hospital admissions, and the extensive need of intensive care units may lead to a figure that is twice as much the need run by other chronic medical conditions [34], adding concerns to its sustainability for even wealthy health-care systems. The apparently never-ending question is: what are we missing, hitherto, in targeting HF outcome?
2.3 Heart Failure Therapy Tomorrow: Looking outside (Beyond) the Current Therapeutic Window
All the therapies that proved to be effective in prolonging HF survival consistently proved to turn down the overexpressed sympathetic-excitatory activity as a primary consequence of pump dysfunction. This is not the only relevant aspect to keep in mind. What we frequently overlook is the pivotal contribution of the autonomic nervous system in maintaining the cardiocirculatory balance by the continuous balancing of its two opposite neuromodulatory systems: the sympathetic or adrenergic system and the parasympathetic or vagal system.
On note, the increased sympathetic activation is coupled to the concomitant, proportional decrease of the counterbalancing vagal nerve activity [35]. This is a critical element for understanding the complex interplay of neurohormonal changes we have learned, since the beta-blocker saga: the autonomic disarray must be stopped and, ideally, reversed.
An intriguing aspect of what we define as autonomic unbalance might reflect the progression of an inherited condition. The findings by Jouven [36] were obtained in a large cohort of persons without history of heart disease and highlighted that the individual heart rate profile during exercise and recovery is an important predictor of sudden death even prior to the time when ischemic heart disease becomes evident and symptomatic. Heart rate responses to exercise are under the control of the autonomic nervous system; these data support the concept that the abnormal response of autonomic balance may precede manifestations of cardiovascular disease and may provide relevant information for early identification of persons at high risk for sudden death.
Data from various studies link increased risk of sudden death to increased sympathetic activity and concomitant decreased vagal activity [36–39]. Very importantly, the autonomic imbalance that marked the population at risk in Jouven’s study is expressed not only by the decreased vagal activity with a higher heart rate at rest and with a lower heart rate recovery but also by lower sympathetic response under effort with an inadequate heart rate increase. Therefore, it means the occurrence of autonomic impairment involves both sides of the system and this is something we did not expect. As addressed by the authors, the association between altered heart rate responses during exercise and sudden cardiac death without associated non-sudden death from myocardial infarction (MI) suggests this risk factor is linked with a specific cardiac arrhythmia susceptibility and it does not reflect the atherosclerotic process. It is consistent with the notion that autonomic imbalance is a predisposing factor to life-threatening arrhythmias beyond the critical contribution of the well-known traditional risk factors. Therefore, it is not surprising that the imbalance, highlighted by the decreased heart rate variability and by the impaired baroreflex response, is a well-recognized indicator of a high risk for sudden death after MI [40], but intriguingly, it becomes a marker of the overall risk of death in HF patients [41] and, despite beta-blocker therapy, can predict overall outcome; the lack of baroreflex sensitivity provides comparable prognosis deterioration even in the treated population [42].
This is a relevant framework of HF that reveals how important is the cardiac substrate in determining the double-edged action of autonomic imbalance on sudden death and on HF death. Thus, the current understanding of autonomic reflex control in HF is that in the early stage of the HF syndrome and as long as the hemodynamic balance is maintained, sympathetic afferent information is the critical determinant of the effective vagal contribution to the autonomic cardiac control. However, at the time when the mechanical deterioration progresses toward the end stage of the syndrome, the humoral adrenergic signaling becomes so prevalent to offset the afferent contribution from the dying heart, leading at the end to affect both modes of HF mortality, sudden and progressive pump failure [39].
It is worth noting that all therapies that proved to be effective on prolonging HF survival restore, to some extent, the baroreceptor competence. This beneficial effect was proved to be present after administration of beta-blocker, after resynchronization therapy and after heart transplantation [42–44]. The finding after heart transplantation is somewhat amazing as the effect is run only by the hemodynamic balance restoration via replacement of the innervated failing heart with a well-performing denervated heart [44]. The conclusion we can draw is that restoration of normal pump performance is able to reset the autonomic system function while autonomic impairment elicited by pump failure can further derange the cardiac performance and the hemodynamic imbalance. Thus, short of replacing the failing heart with a new one, how can we further improve the autonomic imbalance of HF?
It seems reasonable to look at the sympathetic system as the driver of HF disease progression.
Various approaches to modulating the autonomic nervous system have been investigated in order to tap down the excess of sympathetic activation and/or enhance vagal activity. One approach is via renal denervation, which has been hypothesized to decrease the avid sodium and water retention that takes place along the nephron tubule as soon as the kidney flow is decreased in HF. This notion is being tested in ongoing and future research.
Another approach to neuromodulation in HF is to stimulate the peripheral vagal nerve to override the excessive sympathetic drive. The approach has been tested in several studies, but conflicting results have been generated [45–47], addressing the need for more appropriate study design and size and for a better understanding of the “dose ranging” of vagal nerve stimulation. In this regard, with vagal nerve stimulation, it remains a challenge to find the level of appropriate stimulation to recruit efferent fibers with proactive action and contemporaneous recruitment of the afferent vagal component that has inhibitory effect on sympathetic nerve activity [48].
Another perhaps more physiological approach to neuromodulation in HF may be accomplished by stimulating the baroreceptors that are specifically designed to increase vagal output and inhibit the overall sympathetic drive, acting via afferent neural pathway from the baroreceptors to the central nervous system. This approach may allow a truer rebalancing of the autonomic nervous system. These baroreceptors are located in the atrial wall at the junction with pulmonary veins, in the aortic arch, in the glomerular apparatus, and at the bifurcation of carotid vessels in the carotid sinus. They have a common specific action: to turn down sympathetic activity while turning up vagal activity. The application of this approach via an implanted neurostimulator has been termed baroreflex activation therapy (BAT).
Baroreflex activation therapy has been successfully tested in refractory hypertensive patients on top to optimized medical treatment, providing effective blood pressure lowering in long-term follow-up [49]. Of interest, in the treated subpopulation that underwent echocardiographic assessment, positive structural change of the heart has been documented. Specifically, an impressive 18 % reduction of left ventricular mass was seen in association with a highly significant decrease of systolic and diastolic blood pressure [50].
These findings suggest that BAT can be effective in treatment of HF patients, through its effects on the heart and on peripheral mechanism common to hypertension and HF. The possible positive effects of BAT in HF have been demonstrated by the persistence of decreased sympathetic activation in a prolonged follow-up of a small advanced HF population (9 patients) that received BAT. In this study group, muscle sympathetic nerve activity (MSNA) dropped significantly after 3 and 6 months from starting of the therapy and remained unchanged at an average follow-up of 21 months. The MSNA decrease was coupled with a highly significant decrease in the number of days spent by each patient in the hospital in comparison to the one year before BAT [51].
More recently, a randomized controlled trial was completed in 140 NYHA class III reduced ejection fraction HF patients receiving optimal HF drug and electrophysiological device therapies (OMT) alone (N = 69) versus OMT plus BAT (N = 71) [52]. Patients assigned to BAT, compared with control group patients, experienced improvements in the distance walked in 6 min (59.6 ± 14 m vs. 1.5 ± 13.2 m; p = 0.004), quality-of-life score (−17.4 ± 2.8 points vs. 2.1 ± 3.1 points; p <0.001), and NYHA functional class ranking (p = 0.002 for change in distribution). BAT significantly reduced N-terminal pro–brain natriuretic peptide (p = 0.02) and was associated with a trend toward fewer days hospitalized for HF (p = 0.08) (Table 2.1). In addition, BAT significantly increased systolic blood pressure and pulse pressure (Fig. 2.3), correlates of improved survival in HF. Finally, BAT was shown to be safe in this patient population.
Table 2.1




Prominent clinical, biochemical variables changes in baroreflex activation therapy for treatment of heart failure with reduced ejection fraction pivotal study (for details see text) [52]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
