The Renin-Angiotensin System
Barry H. Greenberg
Our understanding of the important role of the reninangiotensin system (RAS) in maintaining circulatory homeostasis and in the pathogenesis of cardiovascular disease has evolved considerably since the late 1890s, when Tigerstedt first observed that aqueous extracts of the kidneys could produce a prolonged increase in blood pressure in an experimental animal model (1). In particular, involvement of the RAS in conditions that lead to the development of heart failure such as hypertension, myocardial infarction (MI), and especially cardiac remodeling, has been well-established for many years now. Moreover, we have come to recognize that the extent, pathways, actions, and interactions ascribed to the RAS are much more varied, far-reaching, and complex than was previously imagined. In this chapter, a description of the RAS and its role in heart failure will be presented. New and emerging pathways and evidence that relates the RAS to the pathogenesis of heart failure will be emphasized. The use of drugs that block RAS activation as a strategy for treating heart failure patients will be discussed briefly and then only in the service of providing evidence that the RAS is involved. Since targeting the RAS has become a cornerstone of heart failure therapy treatment, options will be presented in detail in another chapter of this text.
Description of the Renin-Angiotensin System
As shown in Figure 10-1, initial descriptions of the RAS depicted a cascade of events in which angiotensinogen is released from the liver into the circulation where it is acted upon by renin (generated and released from the juxtaglomerular apparatus of the kidneys) to form the physiologically inactive decapeptide angiotensin I (Ang I). Further degradation of this peptide by angiotensin-converting enzyme (ACE) occurs on the endothelial surface of blood vessels, particularly in the lungs, and results in the formation of Ang II, the main effector molecule of the RAS. Ang II is then distributed in the bloodstream throughout the body where it initiates a wide variety of physiological and pathophysiological effects, most of which are mediated through interactions with its type I (AT1) receptor. Effects of Ang II include arterial vasoconstriction, stimulation of thirst, salt and water retention by the kidney, release of antidiuretic hormone from the posterior pituitary, augmentation of norepinephrine release from sympathetic nerve endings (and inhibition of its reuptake), and remodeling of the heart and blood vessels.
While the circulatory RAS clearly plays an important role in cardiovascular homeostasis, it fails to account for many of the pathophysiological changes that have been ascribed to Ang II during the development and progression of heart failure. For instance, little if any evidence of activation of the circulatory RAS can be detected post-MI once the acute phase is passed (2,3,4). During this quiescent period wherein both plasma renin activity and angiotensin levels are within the normal range, progressive changes in cardiac structure and function that result in the development of heart failure are known to occur. In support of a role of the RAS in this process is a substantial body of evidence from studies done in both experimental animal models and in human patients demonstrating that blocking the generation of Ang II or its interaction with the AT1 receptor can greatly ameliorate the remodeling process (4,5,6,7,8).
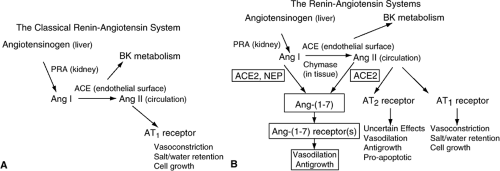 Figure 10-1 Expanding pathways of the renin-angiotensin system (RAS). (A) depicts the classical RAS, while in (B) an expanded version of this system is shown. As discussed in the text, there is information that the tissue-based RAS, the AT2 receptor, and an alternative pathway involving ACE2 all play important roles in regulating cardiac structure and function. |
Discovery that the genes for virtually all components of the RAS are expressed in the heart resulted in the recognition of a tissue-based cardiac RAS (9,10,11,12,13,14). In addition, alternative enzymatic pathways for converting Ang I to Ang II that do not require ACE have been identified. The pathway that has been best-described involves chymase, an enzyme derived from mast cells in the heart (15). There is evidence that the alternative chymase pathway, rather than the one involving ACE, is responsible for the generation of most of the Ang II that is found in human cardiac tissue. The only component of the RAS that may not be generated in sufficient amounts in the heart is renin activity. However, renin from the kidneys can be transported in the bloodstream to the heart, where it is extracted from the coronary circulation (16) and is able to act on locally generated angiotensinogen. The cardiac RAS system is regulated independently of the circulatory system and it functions in an autocrine/paracrine manner to influence cardiac structure and function (10,13,17). In the post-MI setting, substantial upregulation of the cardiac RAS is associated with increases in local concentrations of Ang II (12,18,19). The importance of the cardiac RAS in remodeling is suggested by the close correlation between the extent of structural changes in the heart and cardiac RAS activation (18,19) and by evidence that Ang II levels in the interstitial fluid of the heart are much higher than plasma levels (20).
The use of cardiac-specific transgenic approaches has helped confirm the critical role of the cardiac RAS in the remodeling process (21,22,23). Transgenic mice overexpressing angiotensinogen in cardiomyocytes have been shown to develop cardiac hypertrophy without fibrosis, despite the presence of normal blood pressure. This effect could be inhibited by administration of either an ACE inhibitor or an angiotensin receptor blocker (ARB) (23). Cardiac-specific overexpression of the AT1 receptor results in significant remodeling that is characterized by cardiac hypertrophy and fibrosis (21). Since most effects of Ang II in promoting cardiac remodeling are mediated through its AT1 receptor, the role of the RAS in post-MI cardiac remodeling has been studied in knockout mice that are null for the AT1 receptor. As shown in Figure 10-2, when these mice undergo coronary artery ligation to induce a large MI they develop significantly less left ventricular (LV) dilatation, less fibrosis in noninfarcted segments of myocardium, and better LV systolic function over time than do wild type controls, despite the fact that infarct sizes in the study groups were equal (22). Moreover, post-MI survival is significantly improved in AT1 knockout mice compared to wild type controls.
Effects of Angiotensin II
As previously noted, the AT1 receptor mediates most known effects of the RAS. The mammalian AT1 receptor is a 359-amino-acid protein that has a typical 7-transmembrane domain structure that is similar to other guanine nucleotide regulatory-protein coupled receptors (24,25,26). It has been identified on numerous cell types in tissue throughout the body, including the adrenal gland, heart, kidney, blood vessels, and brain (25,25,26,27,28). In the heart, AT1 receptors are present on cardiac myocytes, fibroblasts, mast cells, and vascular cells (29,30). The AT1 receptor is coupled via heterotrimeric G-proteins to the phospholipase C signal transduction pathway, as evidenced by intracellular calcium mobilization and inositol triphosphate production upon receptor activation (24,31). It has also been linked to transactivation of the epidermal growth factor and other receptor tyrosine kinases and activation of protein kinase C, MAP kinases such as ERK, the JAK/STAT signaling pathway, c-src, and PYK2 kinase (32,33,34,35,36,37,38,39,40,41,42,43). Another important effect of Ang II is activation of the NAD(P)H oxidase, a major source of reactive oxygen species (ROS) production by vascular cells (44).
It is not surprising (given the potential for both local and systemic generation of Ang II, the presence of relevant receptors on numerous cell types throughout the body, and the widely varied signaling pathways that are activated
via the AT1 receptor) that Ang II has substantial effects on the structure and function of numerous organs and organ systems throughout the body. In considering the patho-physiology of heart failure, the effects of Ang II on kidneys, blood vessels, and the heart itself are of greatest interest. Ang II has been associated with many of the conditions that can result in cardiac damage and dysfunction, including hypertension, atherosclerosis, coronary thrombosis, and valvular disease. In the kidney, Ang II promotes the retention of salt and water by both direct effects on the nephron as well as by stimulating the production and release of aldosterone from the adrenal glands. In blood vessels, Ang II has been shown to be a potent vasoconstrictor agent. The renal and vascular effects of Ang II are most important when there is already evidence of cardiac dysfunction. In this setting, salt and water retention predispose to both pulmonary and systemic congestion, while peripheral vasoconstriction reduces delivery of oxygen and nutrients to tissue throughout the body. Moreover, both the salt/water and vascular effects increase load on the heart. While this increase in load may have some favorable short-term benefits (e.g., increasing stroke volume by increasing stretch on cardiac myocytes), these effects are limited and are more than offset by long-term adverse consequences, most notably effects on cardiac remodeling.
via the AT1 receptor) that Ang II has substantial effects on the structure and function of numerous organs and organ systems throughout the body. In considering the patho-physiology of heart failure, the effects of Ang II on kidneys, blood vessels, and the heart itself are of greatest interest. Ang II has been associated with many of the conditions that can result in cardiac damage and dysfunction, including hypertension, atherosclerosis, coronary thrombosis, and valvular disease. In the kidney, Ang II promotes the retention of salt and water by both direct effects on the nephron as well as by stimulating the production and release of aldosterone from the adrenal glands. In blood vessels, Ang II has been shown to be a potent vasoconstrictor agent. The renal and vascular effects of Ang II are most important when there is already evidence of cardiac dysfunction. In this setting, salt and water retention predispose to both pulmonary and systemic congestion, while peripheral vasoconstriction reduces delivery of oxygen and nutrients to tissue throughout the body. Moreover, both the salt/water and vascular effects increase load on the heart. While this increase in load may have some favorable short-term benefits (e.g., increasing stroke volume by increasing stretch on cardiac myocytes), these effects are limited and are more than offset by long-term adverse consequences, most notably effects on cardiac remodeling.
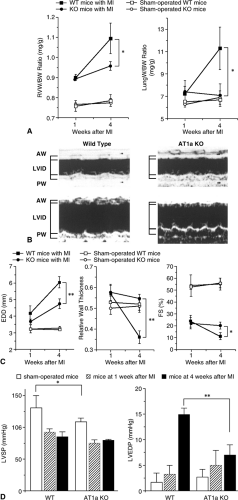 Figure 10-2 Effects of AT1 ablation on post-MI cardiac remodeling. Depicted are echocardiographic and hemodynamic results from AT1 receptor knockout (KO) mice compared to wild type (WT) mice at 1 and 4 weeks post-MI. RVW/BW ratio, right ventricular to body weight ratio; LVID, left ventricular internal dimension; AW, anterior wall; PW, posterior wall. Data expressed as mean ± SEM of 5 to 15 mice per group. *p <0.05; **p 0.01. (From Harada K, Sugaya T, Murakami K, et al. Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation. 1999;100 (20):2093–2099 , with permission.) |
In addition to the indirect effects of the RAS on cardiac remodeling that are mediated by increases in load, it is now recognized that Ang II has direct effects on cells in the heart that promote growth and organ remodeling. Despite the fact that cardiac myocytes have been reported to contain a relatively low density of AT1 receptors (45,46), Ang II (the main effector molecule of the RAS) has been shown to induce cardiac myocyte hypertrophy (30,47,48,49). A possible explanation for this paradox is that Ang II stimulation of other cells that are more richly endowed with AT1 receptors (e.g., cardiac fibroblasts) leads to the generation of numerous secondary growth factors, including interleukin-6 (IL-6), leukemia inhibitory factor-1 (LIF-1), transforming growth factor-beta (TGF-β), and endothelin-1 (ET-1), all of which have been postulated to play a role in promoting cardiac remodeling (46,50,51,52,53,54,55,56,57,58,59). Evidence that Ang II induces hypertrophy of cardiac myocytes only when these cells are co-cultured with cardiac fibroblasts, or when conditioned media from fibroblasts stimulated by Ang II is added to myocyte culture, supports the notion that production of growth factors from fibroblasts is a critical component of the remodeling process (46,51) and that Ang II-mediated cardiac myocyte hypertrophy may not be a direct effect of the peptide. Thus, whether or not Ang II directly causes cardiac myocyte hypertrophy or whether this effect is mediated through the release of secondary growth factors from other cells in the heart is open to question.
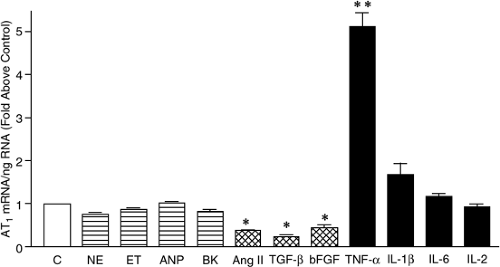 Figure 10-3 AT1 receptor expression in cultured neonatal rat cardiac fibroblasts is increased by proinflammatory cytokines but not by other agents that have been identified in the remodeling heart. *p <0.02; **p <0.01; ***p <0.04. (From Gurantz D, Cowling RT, Villarreal FJ, et al. Tumor necrosis factor-alpha upregulates angiotensin II type 1 receptors on cardiac fibroblasts. Circ Res. 1999;85(3):272–279 , with permission.) |
Ang II stimulates numerous cardiac fibroblast functions that are involved in remodeling. These include replication, migration, and production of extracellular matrix (ECM) proteins and secondary growth factors. Most of these effects are mediated through the AT1 receptor (31,60,61,62,63,64), which is considerably more abundant on cardiac fibroblasts than on cardiac myocytes (45). Furthermore, in pathological settings in which cardiac remodeling occurs (e.g., following an MI), the density of the AT1 receptor on cardiac fibroblasts is increased (65,66,67,68). As shown in Figure 10-3, the proinflammatory cytokines, tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β) have been shown to be potent inducers of AT1 receptor upregulation in cultured cardiac fibroblasts, whereas other growth factors known to be present in the remodeling heart either fail to increase or actually decrease AT1 receptor expression (69). Proinflammatory cytokine-mediated AT1 receptor upregulation involves activation of the transcription factor nuclear factor-kappa B (NF-KB) since it can be selectively blocked by interfering with NF-KB dissociation from IKB, a regulatory protein that restricts nuclear translocation which is essential to activate gene expression (70).
In the post-MI heart there are both temporal and spatial associations between increases in AT1 receptor density and the appearance of these proinflammatory cytokines that are consistent with the possibility that the relationship may be causal in nature (71). In addition, receptor upregulation induced by TNF-α and IL-1β appear to be additive. Although the significance of increased AT1 receptor density on cardiac fibroblasts during remodeling is uncertain, there is evidence that proinflammatory cytokine-induced AT1 upregulation enhances Ang II-stimulated proline incorporation (depicted in Figure 10-4) and production of
TIMP-1 (72). These findings strongly suggest that upregulation of the AT1 receptor contributes to the remodeling process by increasing the responsiveness of cardiac fibroblasts to the profibrotic effects of Ang II (69,72).
TIMP-1 (72). These findings strongly suggest that upregulation of the AT1 receptor contributes to the remodeling process by increasing the responsiveness of cardiac fibroblasts to the profibrotic effects of Ang II (69,72).
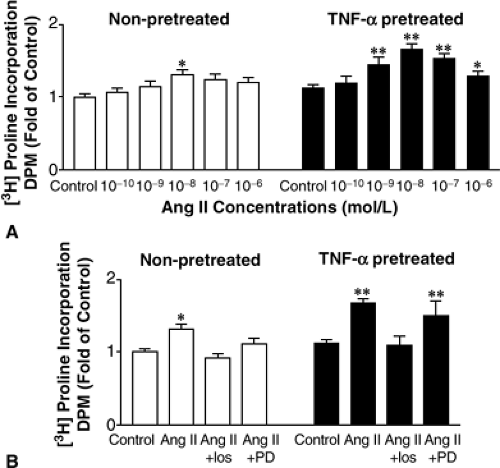 Figure 10-4 (A) Ang II-stimulated [(3)H]proline incorporation is enhanced by TNF-α pretreatment. In nonpretreated fibroblasts (open bars), Ang II-stimulated [(3)H]proline incorporation throughout a concentration range of 10-10 to 10-6 mol/L. Pretreatment of the cells with TNF-α (10 ng/mL) for 48 hours, however, enhanced the profile of [(3)H]proline incorporation by Ang II (filled bars). (B) Effect of losartan and PD123319 (both at 10-(6) mol/L) on [(3)H]proline incorporation by Ang II (10-8 mol/L. Losartan (Los), the AT1 receptor antagonist, but not PD123319 (PD), the AT2 receptor antagonist, blocked the Ang II effects in both nonpretreated (open bars) and TNF-α-pretreated (filled bars) cells. Data are presented as mean ± SEM fold of control. For (A) and (B), *p <0.05, **p <0.01, n = 7 to 8). (From Peng J, Gurantz D, Tran V, et al. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91(12):1119–1126 , with permission.) |
As previously noted, most Ang II effects are mediated by the AT1 receptor. It is now recognized, however, that Ang II can also bind to its type 2 (AT2) receptor with effects that may have important consequences in the cardiovascular system (Fig. 10-1B). Although both receptor subtypes are 7-transmembrane domain G-protein-coupled receptors of the class A rhodopsin-like family, there is only ∼30% sequence homology between them (73,74). Both the AT1 and the AT2 receptors have affinities for Ang II that are in the nanomolar range. The AT2 receptor is highly expressed in developing fetuses (27,75). After birth, its expression declines rapidly and in the adult it is expressed in very low abundance in the cardiovascular system. Under pathological, conditions, however, expression levels may increase substantially (76,77,78). Signaling pathways associated with the AT2 receptor include activation of protein phosphatases (with resultant protein dephosphorylation), activation of phospholipase A2, regulation of the bradykinin-NO-cyclic guanosine monophosphate (cGMP) system, and sphingolipid-derived ceramide formation (79,80,81,82). Activation of the AT2 receptor has been associated with an increased rate of cell apoptosis (83).
The role of the AT2 receptor in cardiovascular home-ostasis and, particularly, in remodeling of the heart has not yet been fully delineated. Consistent with evidence indicating an association between the AT2 receptor and protein phosphatase activity, initial reports of the effects of AT2 receptor activation suggested that it mediated antigrowth effects that opposed those of the AT1 receptor in cardiac myocytes and fibroblasts (84,85,86,87). However, some experiments in mice that were null for the AT2 receptor gene have provided conflicting results in regard to its impact on the remodeling process. For instance, Ichihara et al. found that the chronic loss of the AT2 receptor abolished the development of LV hypertrophy and cardiac fibrosis in chronic Ang II-induced hypertension, suggesting the necessity of this receptor subtype for the development of these pathological structural changes (88). Of interest is the observation that transgenic mice with ventricular-specific increases in AT2 receptor density develop a phenotype of dilated cardiomyopathy characterized by ventricular dilatation, wall thinning, increased fibrosis, and depressed contractile function (89). Although the mechanism for these abnormalities in ventricular structure and function are uncertain, increased apoptosis within the ventricular myocardium may be involved. In support of this possibility was the observation that the severity of the phenotype and extent of apoptosis were directly related to the level of expression of AT2 receptor gene expression (83). Furthermore, the transgenic animals with cardiac-specific AT2 receptor overexpression also demonstrated evidence of increased levels of activation of PKC-α and -β, both of which have been implicated in cardiac remodeling and the development of heart failure.
Complicating this picture is evidence that the AT2 receptor might influence the response to Ang II through formation of structural heterodimers with the AT1 recepto. In this case the interposition of the AT2 molecule into the usual AT1 homodimer has been shown to inhibit AT1 receptor signaling. Thus, AT2 molecules could serve a dominant-negative function and antagonize the AT1 receptor (84). Since both of these Ang receptor subtypes are coexpressed in many tissues, the precise effects of Ang II signaling might well be related to the relative abundance of the two receptor subtypes. Thus, in settings where the AT2 receptor is downregulated, the ratio between the receptors would result in a lower likelihood of heterodimerization, an effect that would tend to favor an enhanced response of the AT1 receptor to Ang II. Conversely, situations that result in an increase in the ratio of AT2 to AT1 would be expected to decrease AT1-mediated effects of Ang II.
A role for the AT2 receptor in the development of cardiac hypertrophy in humans was suggested by the results of a recent study evaluating the association between a common intronic polymorphism of the AT2 gene (-1332 G/A) and the presence of LV hypertrophy in hypertensive patients (90). The AT2 gene consists of 3 exons and 2 introns, with the entire reading frame for the AT2 receptor located on exon 3. The polymorphism site is located just downstream from exon 2 in a region that is important for transcriptional activity. Individuals with the G allele lack exon 2 of the AT2 gene; this leads to less-effective transcription. The fact that there was an association between the G allele and LV hypertrophy (despite the fact that patients
had been treated for their hypertension) is consistent with the possibility that the AT2 receptor may mediate antihypertrophic effects in this setting. Clearly, the role of the AT2 receptor in cardiac remodeling and heart failure will require further study in order to determine its importance.
had been treated for their hypertension) is consistent with the possibility that the AT2 receptor may mediate antihypertrophic effects in this setting. Clearly, the role of the AT2 receptor in cardiac remodeling and heart failure will require further study in order to determine its importance.
The Angiotensin-Converting Enzyme-2 Pathway
Recently, an alternative pathway of the RAS involving a homolog of ACE, termed ACE2, has been described (91,92) and there is emerging evidence that it may play an important role in the cardiovascular system (93). Human ACE2 cDNA predicts an 805-amino-acid protein that has a 42% homology with the N-terminal catalytic domain of ACE (91). Although first identified in the heart, kidney, and testes (91,94), the distribution of ACE2 now appears to be considerably more widespread than was originally believed (92).
As shown in Figure 10-1B, ACE2 functions as a carboxypeptidase that cleaves the terminal amino acid from either the decapeptide Ang I to form Ang-(1,2,3,4,5,6,7,8,9) or the octapeptide Ang II to form Ang-(1,2,3,4,5,6,7). The catalytic activity of ACE2 for Ang II is substantially greater than for Ang I, suggesting that its primary role may be in the conversion of Ang II to Ang-(1,2,3,4,5,6,7) (95). Unlike ACE activity, ACE2 is neither able to convert Ang I to Ang II nor break down bradykinin (91,95). Of particular importance is the fact that the activity of ACE2 is not affected by ACE inhibitors (91,94). ACE2 has also been found to be a functional receptor for the severe acute respiratory syndrome coronavirus (SARS-CoV) (96) and there is evidence that the Spike protein of the virus reduces ACE2 expression, an effect that has been postulated to be related to the virulence of the pneumonia caused by this agent.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


