The Peripheral Circulation in Heart Failure
Anju Nohria
Mark A. Creager
The development and progression of myocardial failure are heralded by the activation of circulating neurohormonal systems that modulate both vascular tone and renal retention of salt and water. The peripheral circulation undergoes local changes in response to heart failure that are fundamental to the pathophysiology of this disease state. The fractional distribution of blood flow to the kidneys, limbs, and splanchnic beds decreases, whereas blood flow to the heart and brain is preserved (1,2). The diminished exercise capacity of limb muscles in patients with heart failure may be due in part to chronically diminished nutritive perfusion (3,4). Renal hypoperfusion and altered intrarenal hemodynamics may contribute to sodium and water retention (5,6). Previous chapters have focused on the mechanisms underlying systemic activation of the sympathetic nervous system, the renin-angiotensin system, as well as other circulatory neurohormones, such as arginine vaso-pressin and natriuretic peptides. In this chapter we will examine the structural and functional changes that occur in the peripheral vasculature. Specifically, this chapter will focus on local vasodilator and vasoconstrictor mechanisms that are mediated by the endothelium, by tissue production of angiotensin II, and by adrenergic and vasopressinergic receptors. The vascular effects of endogenous vasodilators, including the natriuretic peptides and adrenomedullin, will be discussed as well.
Endothelial Mechanisms in Heart Failure
The response of large arteries and arterioles to both physiological and pharmacological stimuli is influenced by the presence of an intact endothelium. Vascular endnthelial cells have been shown to synthesize endogenous vasodilators (e.g., nitric oxide and prostacyclin) as well as vasoconstrictors (e.g., endothelin and vasoconstrictor prostanoids). Regulation of endothelium-derived vasodilation and constriction may be altered in heart failure and contribute to the vasoconstricted state (Table 14-1). The data supporting this contention are reviewed next.
Nitric Oxide
Furchgott and Zawadzki initially demonstrated that relaxation of vascular rings in response to acetylcholine was dependent on an intact endothelium and was abolished by removal of the endothelium (7). This relaxation is due to the release of an endogenous substance called nitric oxide (8,9,10). Nitric oxide elicits vascular smooth muscle dilation by activating soluble guanylyl cyclase, thereby increasing intracellular levels of cyclic guanosine monophosphate (cGMP).
Nitric oxide is the most potent endogenous vasodilator yet identified and contributes to the regulation of basal vascular tone. Nitric oxide is synthesized by nitric oxide synthase from the amino acid, L-arginine (11,12) in response to a wide variety of stimuli (Fig. 14-1). A number of vasoconstrictors, such as norepinephrine, vasopressin, thrombin, and endothelin, interact with specific receptors on the endothelial surface to induce the release of nitric oxide, thus modulating the direct vasoconstrictor effect of these substances. Aggregating platelets also release vasoactive substances (e.g., adenosine diphosphate and serotonin) that stimulate nitric oxide production and thereby prevent further platelet aggregation. Thus, in the presence of an intact endothelium, nitric oxide counterbalances the deleterious effects
of vasoconstrictive and thrombogenic stimuli. However, after endothelial cell injury, the predominant effects of these stimuli are mediated by their receptors on vascular smooth muscle, activation of which causes vasoconstriction (13,14,15). Thus, alterations in the bioavailability of nitric oxide in disease states might lead to increased vascular resistance and may be associated with changes in vascular structure and increased thrombosis.
of vasoconstrictive and thrombogenic stimuli. However, after endothelial cell injury, the predominant effects of these stimuli are mediated by their receptors on vascular smooth muscle, activation of which causes vasoconstriction (13,14,15). Thus, alterations in the bioavailability of nitric oxide in disease states might lead to increased vascular resistance and may be associated with changes in vascular structure and increased thrombosis.
Table 14-1 Effects of Endogenous Vasoactive Substances in Heart Failure | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
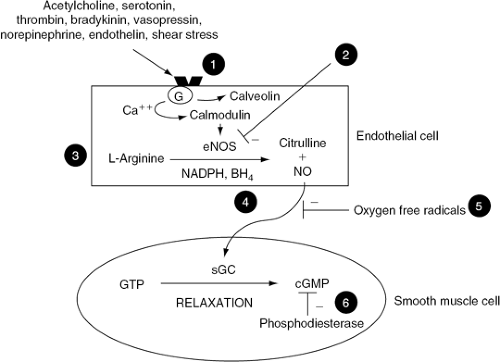 Figure 14-1 Endothelial cell production of nitric oxide (NO) and the potential mechanisms for endothelial dysfunction in heart failure, Stimulation of endothelial cells leads to an increase in intracellular calcium (Ca2+), displacing calveolin from calmodulin, activating endothelial nitric oxide synthase (eNOS). eNOS synthesizes NO from L-arginine in the presence of several cofactors, including NADPH and tetrahydrobiopterin (BH4). NO diffuses to the underlying vascular smooth muscle and causes vasodilation by activating soluble guanylate cyclase (sGC), thereby increasing intracellular cyclic guanosine monophosphate (cGMP). Heart failure reduces the bioavailability of NO by several potential mechanisms: (1) uncoupling of endothelial receptors from associated G-proteins needed for signal transduction; (2) decreasing synthesis and activity of eNOS; (3) reducing substrate availability for the synthesis of NO; (4) reducing cofactors necessary for the activity of eNOS; (5) degradation of NO by oxygen free radicals; and (6) inactivation of cGMP by increased phosphodiesterase activity. |
Nitric Oxide in Heart Failure
Endothelium-dependent vasodilation is attenuated in animal models of heart failure. Vasodilator responses to acetylcholine, but not to nitroglycerin, are diminished in the peripheral and coronary arteries of dogs with pacing-induced heart failure (16,17). Moreover, inhibition of basal nitric oxide biosynthesis by the administration of L-N mono methyl arginine (L-NMMA), an inhibitor of nitric oxide synthase, results in a marked increase in mean arterial pressure in conscious healthy dogs but not in dogs with pacing-induced heart failure (18). These results suggest that basal release of nitric oxide is reduced in this model of heart failure.
A number of studies have shown that endothelium-dependent vasodilation is also abnormal in humans with heart failure. Acetylcholine-mediated coronary vasodilation is diminished in patients with idiopathic dilated cardiomy-opathy (19). Several groups of investigators have also evaluated the responsiveness of the forearm circulation to endothelium-dependent and endothelium-independent vasodilators (20,21,22). The increase in forearm blood flow during intrabrachial artery infusion of acetylcholine and methacholine, both endothelium-dependent vasodilators, is blunted in patients with heart failure, whereas forearm vascular responsiveness to the endothelium-independent vasodilator, nitroprusside, is preserved (Fig. 14-2). Acetylcholine-induced changes in brachial artery diameter, a measure of peripheral conduit vessel dilation, are also abnormal in patients with heart failure (20). The blunted forearm blood flow response to acetylcholine is not associated with duration of symptoms or abnormal hemodynamic indexes; rather, it correlates with maximal oxygen consumption at peak exercise (23). Hence, it is not surprising that endothelial dysfunction is an early predictor of mortality in patients with heart failure (24,25).
The exact mechanism contributing to the decreased bioavailability of nitric oxide in patients with heart failure remains unclear. Decreased production of nitric oxide might contribute to endothelial dysfunction in heart failure (Fig. 14-1). Endothelial cells produce nitric oxide from L-arginine via nitric oxide synthase. It has been suggested that a reduction in the expression of cationic amino acid transporters and thus arginine transport by endothelial cells may lead to a relative deficiency of intracellular arginine, affecting nitric oxide synthesis (26). This may provide one explanation for impaired nitric oxide synthesis and for the beneficial response to acetylcholine seen after the intravenous (27) and oral administration of L-arginine in patients with heart failure (28). A decrease in the bioavailability of tetrahydrobiopterin, an essential cofactor of nitric oxide synthase, may also contribute to decreased nitric oxide production since infusion of tetrahydrobiopterin improves acetylcholine mediated forearm vasodilation in patients with heart failure (29). Conversely, increased degradation of nitric oxide or its downstream effectors might also contribute to endothelial dysfunction in heart failure (Fig. 14-1). Increased oxidative stress and nitric oxide inactivation by superoxide anions have been proposed as an additional mechanisms contributing to endothelial dysfunction in heart failure.
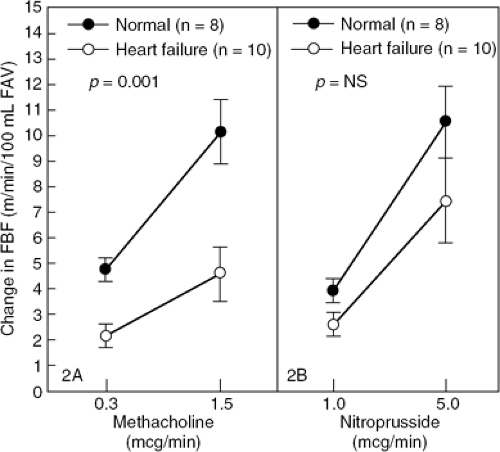 Figure 14-2 The vascular effects of intrabrachial infusion of an endothelium-dependent vasodilator (methacholine) and an endothelium-independent vasodilator (nitroprusside) in normal subjects and in patients with heart failure. The methacholine dose response was blunted in patients with heart failure (left panel); in contrast, the nitroprusside dose response was not significantly decreased in heart failure patients. FBF, forearm blood flow. (Adapted from Kubo SH, Rector TS, Bank AJ, et al. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation. 1991;84:1589–1596 .) |
The three enzyme systems responsible for free radical production within the vessel wall include nicotinamide adenine dinucleotide/nicotinamide adenine dinucleotide phosphate (NADH/NADPH) oxidase, xanthine oxidoreductase, and endothelial nitric oxide synthase (30). Strategies aimed at preventing superoxide anion formation, such as inhibition of NADPH oxidase by statins (31) and xanthine oxidoreductase by allopurinol (32,33), have been shown to improve the forearm blood flow response to acetylcholine in patients with heart failure. Furthermore, neutralization of the harmful effects of superoxide anions by the antioxidant vitamin C improves flow-dependent vasodilation in chronic heart failure (34). Exercise training improves endothelial function and is associated with a 100% increase in the transcript levels of genes encoding the antioxidant enzymes, copper zinc superoxide dismutase and glutathione peroxidase (35). Reduction in oxidant stress may be one mechanism to explain the improvement in endothelial function associated with exercise training in patients with heart failure (36).
Nitric oxide mediates its effects via cGMP. Increased degradation of cGMP by phosphodiestereases, in particular
type 5 phosphodiesterase, might also lead to increased vasoconstriction in heart failure. Inhibition of type 5 phos-phodiesterase with sildenafil has also been shown to increase flow-mediated, endothelium-dependent vasodilation in patients with heart failure (37). The relative contribution of these potential mechanisms for decreased nitric oxide bioavailability in heart failure remains unclear.
type 5 phosphodiesterase, might also lead to increased vasoconstriction in heart failure. Inhibition of type 5 phos-phodiesterase with sildenafil has also been shown to increase flow-mediated, endothelium-dependent vasodilation in patients with heart failure (37). The relative contribution of these potential mechanisms for decreased nitric oxide bioavailability in heart failure remains unclear.
Many patients with heart failure have other coexisting diseases, such as atherosclerosis. It is well-established that endothelium-dependent vasodilation is abnormal in atherosclerotic vessels (38). Endothelial function is impaired in animals and humans with risk factors for atherosclerosis, such as hypercholesterolemia, hypertension, hyperhomocysteinemia, and diabetes, even in vessels without overt atherosclerotic lesions (39). Therefore, it is conceivable that abnormalities in endothelium-dependent vasodilation in some patients with heart failure reflect the presence of these other disease states, in addition to the effect of heart failure alone.
Prostaglandins
Endothelial cells synthesize the vasodilator prostaglandins E2 and I2 (PGE2 and PGI2) from arachidonic acid. Increased systemic levels of vasodilator prostaglandins are observed in patients with heart failure. In patients with severe left ventricular dysfunction, the prostaglandin metabolites of PGE2 and PGI2 are three- to tenfold higher than levels measured in normal subjects (40). The mechanisms underlying increased prostaglandin levels in heart failure are not known. Local synthesis of vasoactive prostanoids is increased by hypoperfusion of regional vascular beds (e.g., the renal and coronary circulations [41,42] and production of prostaglandins may directly be stimulated by vasoconstrictor hormones, such as angiotensin, vasopressin, and norepinephrine [43–45]). The concentration of prostanoids is not increased in all patients, but levels of these vasodilator prostaglandins correlate directly with the concentrations of angiotensin II and plasma renin activity. Thus, it appears that synthesis of prostaglandins by blood vessels counterbalances vasoconstrictor mechanisms in heart failure.
The contribution of local vasodilator prostaglandins to vascular resistance has important clinical implications. Prostaglandin-synthetase inhibition in patients with heart failure causes a decline in cardiac output and an increase in pulmonary capillary wedge pressure, particularly in those subjects with the greatest activation of circulating vasoconstrictor hormones (40). Additionally, some pharmacological agents produce their beneficial effects in part by increasing the production of endogenous vasodilator prostaglandins. The vasodilation induced by captopril, nitroglycerin, nitroprusside, and hydralazine is attenuated by pretreatment with indomethacin (46,47,48). In addition to its effects on angiotensin, captopril inhibits the breakdown of bradykinin by kininase II. Bradykinin may be an important contributor to the short-term vasodilatory effect of captopril (49), potentially by stimulating release of prostacyclin (50). Acetylsalicylic acid, another inhibitor of cyclooxygenase, has been found to blunt the augmentation in endothelium-dependent vasodilation induced by enalaprilat in patients with mild heart failure, highlighting the contribution of prostaglandins to the peripheral vascular effects of angiotensin-converting enzyme (ACE) inhibitors in this population (51,52).
Endothelin
The endothelium also produces potent vasoconstrictor substances. Yanagisawa et al. identified the endothelium-derived vasoconstrictor, endothelin, in 1988 (53). Endothelin, a 21-amino acid peptide hormone, is the most potent endogenous vasoconstrictor substance identified to date. Endothelin is synthesized from its messenger RNA (mRNA) in endothelial cells as a pre-propeptide and is cleaved to big endothelin by an endogenous endopeptidase. Mature endothelin is then produced by an endothelin-specific converting enzyme, which is present in the lung and in other endothelial cells (54). Many of the same stimuli that increase release of nitric oxide (including shear stress, epinephrine, thrombin, angiotensin II, arginine vasopressin, and calcium ionophore) also increase transcription of the pre-proendothelin mRNA and increase release of endothelin (53,55,56,57,58).
Three isoforms of endothelin have been identified and sequenced: endothelin-1 (ET-1), ET-2, and ET-3. The ET-1 isoform is most widely associated with cardiovascular regulation. ET-1 acts through the endothelin A (ETA) and endothelin B (ETB) receptor subtypes. The ETA receptor, found on vascular smooth muscle cells, mediates vasoconstriction as well as smooth muscle cell proliferation (53,59) (Fig. 14-3). ETB is present on both vascular smooth muscle cells and endothelial cells, where it mediates ET-1-induced vasoconstriction and vasodilation, respectively (60,61) (Fig. 14-3).
Endothelin binds to these specific receptors and activates several intracellular signal transduction systems via guanosine triphosphate (GTP)-binding proteins. Endothelin stimulates phospholipase C (PLC) and opens voltage-dependent calcium channels. Activation of PLC degrades phosphoinositide to form inositol triphosphate (IP3) and diacylglycerol. IP3 releases calcium from intracellular stores, and diacylglycerol sensitizes contractile proteins via protein kinase C (62,63,64,65). Endothelin may also promote vasoconstriction by upregulation of the endogenous nitric oxide synthase inhibitor, asymmetric dimethyl-arginine, thus reducing nitric oxide synthesis (66). Endothelin also stimulates phospholipase A2 to produce several vasoconstrictor prostanoids (62,64). The effects of endothelin are most prominent in adjacent vascular regions and, thus, endothelin serves as an autocoid modulator of arteriolar resistance.
The vascular effects of endothelin are complex and potentially clinically important. During exogenous infusion of endothelin there is an initial, brief vasodilation followed by prolonged vasoconstriction (60,61,67,68,69). Infusion of exogenous endothelin into normal animals increases systemic vascular resistance and consequently reduces cardiac output via cardiac and associated peripheral vascular effects (67,68,70). Chronic exposure of vascular smooth muscle cells to endothelin has been shown to cause proliferation in vitro (71,72,73). Like peptide growth factors, endothelin mediates cellular growth via tyrosine kinases and mitogen-activated protein kinase pathways (74).
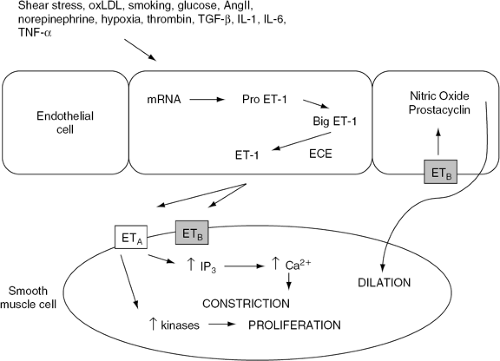 Figure 14-3 Synthesis and effects of endothelin. In response to several stimuli, endothelin (ET-1) is synthesized from its messenger RNA (mRNA) as a prepropeptide (pro ET-1) and is cleaved to big endotnelin (Big ET-1) by an endogenous endopeptidase. Mature ET-1 is then produced by endothelin converting enzyme (ECE). ET-1 acts on endothein A (ETA) receptors on vascular smooth mussle cells to activate phospholipase C that degrades phos-phoinositide to form inositol triphos-phate (IP3) and diacylglycerol. IP3 releases calcium (Ca2+) from intracellular stores to cause contraction. ET-1 also activates (ETA) receptors to mediate cellular growth via tyrosine kinase and mitogen-activated protein kinase pathways. ET-1 also activates the endothelin B (ETB) receptor present on vascular smooth muscle cells to mediate vaso-constriction. However, the ETB receptor on endothelial cells promotes the release of nitric oxide (NO) and stimulates phospholipase A2 to produce prostanoids that mediate vasodilation. |
Endothelin in Heart Failure
A two- to threefold increase in plasma concentrations of endothelin has been observed in experimental heart failure induced by rapid ventricular pacing in the dog (75,76). Elevations in plasma endothelin concentration also occur in humans with stable or decompensated heart failure (76,77,78,79,80).
Plasma endothelin levels may be increased in heart failure by several mechanisms. Catecholamines, angiotensin II, and arginine vasopressin stimulate endothelin synthesis (53,56,81) and endothelin clearance also is decreased in heart failure. The pathophysiological significance of increased plasma endothelin levels has become more apparent with the advent of endothelin receptor antagonists. In a rat model of post-myocardial infarction heart failure, both ETA and combined ETA/B receptor antagonism improved acetylcholine-mediated vasodilation and reduced superoxide anion formation (82) (Fig. 14-4). Similarly, treatment with the ETA receptor blocker, LU135252, improved flow-mediated vasodilation in the brachial artery of humans with heart failure (83). Blockade of endogenous endothelin with a short-term infusion of the nonselective antagonist bosentan, lowered ventricular filling pressures and systemic vascular resistance in patients with heart failure (84). However, the promise of both specific and combined endothelin receptor antagonists has not been realized in large clinical trials, suggesting that the effects and importance of the two endothelin receptors are poorly understood (85,86,87) (Table 14-2).
Renin-Angiotensin System
The role of the circulating renin-angiotensin system in the maintenance of cardiovascular homeostasis has been defined by use of specific antagonists. Antirenin antibodies (88), angiotensin II antagonists (89), and ACE inhibitors (90) have all been used in this context. Studies with these agents have demonstrated that acute hemodynamic responses correlate with pretreatment plasma renin-angiotensin system activity, and are most profound in individuals with high plasma renin activity and angiotensin II concentrations. In contrast, chronic responses to these agents are not predicted by plasma renin-angiotensin system activity. Indeed, ACE inhibition is also effective in heart failure patients with normal plasma renin activity (91,92). Therefore, circulating angiotensin II may not account wholly for angiotensin II-mediated vasoconstriction in heart failure.
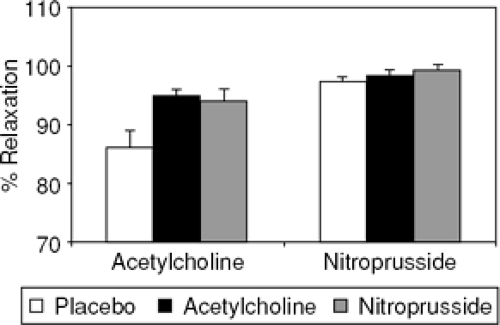 Figure 14-4 Acetylcholine (endothelium-dependent)- and nitro-prusside (endothelium-independent)- induced relaxation of phenylephrine preconstricted aortic rings from rats with heart failure after myocardial infarction. Both the endothelin A specific receptor antagonist, LU135252, and the nonspecific endothelin A and B receptor antagonist, bosentan, improved acetylcholine-mediated relaxation relative to placebo. These data suggest that endothelin antagonists improve nitric oxide bioavailability in blood vessels from animal models of heart failure. (Adapted from Bauersachs J, Fraccarollo D, Galuppo P, et al. Endothelin-receptor blockade improves endothelial vasomotor dysfunction in heart failure. Cardiovasc Res. 2000;47:142–149 .) |
Table 14-2 Selected Randomized Clinical Trials of Endothelin Antagonists in Heart Failure | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Endogenous renin-angiotensin systems have been demonstrated in tissues that are important in cardiovascular regulation (e.g., blood vessels, heart, kidney, brain, and adrenal tissues). The evidence supporting the existence and physiological role of vascular and renal renin-angiotensin systems is reviewed below. The possibility that the tissue renin-angiotensin system might contribute to local angiotensin II production has important pathophysiological and therapeutic implications.
The Vascular Renin-Angiotensin System
The presence of renin, angiotensinogen, and angiotensin II in blood vessels has been reported by a number of laboratories (93,94). The vessel wall distribution of renin has been examined by use of antirenin-specific antibody, and intense staining has been noted throughout the thickness of the aorta, large and smaller arteries, as well as arterioles, particularly in endothelial and smooth muscle cells (95). Additionally, kinetic analysis of arterial-venous angiotensin I and angiotensin II differences in humans indicates that both angiotensin I and angiotensin II are synthesized in vascular tissues (96).
The local synthesis of angiotensin II in the blood vessel wall has important physiological implications. Angiotensin II has multiple effects on the cardiovascular system, which include not only vasoconstriction, but also smooth muscle cell proliferation, myocardial hypertrophy, and altered ventricular remodeling (97). Conceptually, local increases in vascular angiotensin II can cause constriction of large arteries and resistance vessels via the angiotensin type I receptor, resulting in increased systemic vascular resistance and reduced arterial compliance. Local angiotensin II also may contribute directly to regional blood flow regulation by activating vascular receptors in specific circulations (e.g., the kidney). Angiotensin II receptors are also found on endothelial cells, and activation can result in release of both nitric oxide (98) and vasoconstrictor prostanoids (99). Angiotensin II also may alter vascular function by facilitating norepinephrine release from local noradrenergic nerve terminals (100). In addition to these mechanisms, there is data suggesting that angiotensin II activates membrane-bound NADH/NADPH-driven oxidases that promote superoxide anion generation in cultured vascular smooth muscle cells (101). Cells exposed to angiotensin II for 4 hours produce two- to sevenfold more superoxide anion than control cells. This increase can be reversed by co-incubation with an angiotensin II receptor antagonist. Angiotensin II-induced, but not norepinephrine-induced, hypertension is associated with impaired vascular response to acetylcholine (102). Treatment with the angiotensin II receptor antagonist, losartan, improves the response to acetylcholine and normalizes vascular superoxide production, implicating the angiotensin type-1 receptor as a mediator in this process. These autocoid mechanisms may affect regional vascular tone, even when circulating levels of angiotensin II are not increased.
Evidence to support the importance of local angiotensin II to arteriolar and conduit artery function is derived from experiments involving ACE inhibition or angiotensin receptor blockade. Local infusion of ACE inhibitors to humans causes limb and coronary vasodilation, indicating that local generation of angiotensin II may contribute to vascular resistance (103). Intra-arterial infusion of quinapril augments the forearm blood flow response to bradykinin. The increase in arterial flow caused by quinapril can be inhibited by L-NMMA, suggesting that this ACE inhibitor may increase vascular nitric oxide, perhaps through a bradykinin-mediated pathway (104). The compliance and diameter of large (brachial and carotid) arteries are increased by ACE inhibition, even at doses that do not reduce blood pressure (105,106). This latter observation suggests that the local vascular renin-angiotensin system activity influences distensibility of these conduit vessels.
The Renal Renin-Angiotensin System
The juxtaglomerular apparatus is capable of releasing large amounts of renin into the circulation. In patients with heart failure, low renal perfusion pressure, decreased distal tubular sodium load, increased sympathetic activity, and diuretic administration all increase systemic levels of renin and angiotensin II. The presence of a locally active renin-angiotensin system in the kidney, in sites other than the juxtaglomerular cells, has been documented using molecular biological, immunocytochemical, and biochemical techniques. Renin has been demonstrated in afferent and efferent arterioles and in the proximal tubule by antirenin antiserum staining (107,108). Cultured glomerular mesangial cells synthesize renin (93). Angiotensinogen mRNA expression in the renal cortex has been demonstrated and in situ hybridization studies have
shown that angiotensinogen mRNA is expressed principally in the proximal tubule (109,110), while renin mRNA is primarily localized to the juxtaglomerular cells (110). Intrarenal ACE also has been demonstrated by the presence of ACE mRNA (111). In addition to the vasculature, ACE has been localized to the proximal tubule brush border using immunohistochemical and radioligand binding techniques (112). Because all the components are found in the proximal tubule, local synthesis of angiotensin II has been hypothesized (110). Indeed, a micropuncture study demonstrated that proximal tubular fluid angiotensin II concentration is 1,000-fold greater than in the plasma (113). Local angiotensin II production might be a major factor in regulation of basal renal hemodynamics and sodium reabsorption.
shown that angiotensinogen mRNA is expressed principally in the proximal tubule (109,110), while renin mRNA is primarily localized to the juxtaglomerular cells (110). Intrarenal ACE also has been demonstrated by the presence of ACE mRNA (111). In addition to the vasculature, ACE has been localized to the proximal tubule brush border using immunohistochemical and radioligand binding techniques (112). Because all the components are found in the proximal tubule, local synthesis of angiotensin II has been hypothesized (110). Indeed, a micropuncture study demonstrated that proximal tubular fluid angiotensin II concentration is 1,000-fold greater than in the plasma (113). Local angiotensin II production might be a major factor in regulation of basal renal hemodynamics and sodium reabsorption.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


