Figure 1.1
Factors involved in the pathophysiology of pulmonary arterial hypertension
Vasoconstriction
In accordance with Poiseuille’s law the flow of blood through a vessel is proportional to its radius to the fourth power. Consequently, relatively small reductions in vessel radius are accompanied by large reductions in blood flow. For example if the radius of a vessel is halved then blood flow is decreased by a factor of 16. Paralleling Ohm’s law relating to electrical principles, PVR is determined by dividing the pressure drop through the pulmonary circulation by total pulmonary blood flow. Therefore a significant fall in blood flow from a reduction in the radius of vessels is associated with a significant increase in PVR.
Cellular Proliferation and Thrombosis
In the early stages of PPA hypertrophy, recruitment and proliferation of smooth muscle results in thickening of the subendothelial tunica media layer of the distal pulmonary arteries. Activated vascular cells then obtain migratory and invasive properties in addition to hyperproliferative properties. They migrate from the inner half of the tunica media of pulmonary arterioles and travel to the vascular lumen. In the lumen they become myofibroblasts which proliferate and lay down smooth muscle and fibrous tissue in a concentric fashion. This causes vascular narrowing (i.e., a reduction in vessel radius) and/or occlusion and blood flow is significantly reduced according to Poiseuille’s law so PVR increases. At points of weakness, especially when the blood vessels are branching, they rupture with subsequent haemorrhage. Disorganised primitive blood vessels grow into these areas and are termed plexiform lesions. The plexiform lesions are composed of multiple cell types including apoptosis-resistant endothelial cells, progenitor cells and immune cells [3, 7].
The endothelial dysfunction and the associated imbalance in endothelium derived vascular mediators gives rise to a hypercoagulable phenotype in PAH [8]. This promotes the development of in situ thrombosis. The development of thrombus in the already narrowed pulmonary arteries further obstructs and reduces blood flow, increasing PVR.
Mechanisms of Disease
The exact mechanisms behind the development of PPA are not fully understood. Pulmonary endothelial dysfunction has a central role in its initiation and progression [9–12]. Certain risk factors can facilitate this endothelial damage and dysfunction, including autoimmune diseases, toxins and HIV infection. Inflammation, including cytokines and immune cells appear to play a significant role in the initiation and evolution of PPA [13]. Many factors with roles in normal cell growth and/or angiogenesis are altered to promote cell proliferation and disordered angiogenesis [14–16]. Mitochondrial metabolism in proliferating vascular cells in PPA is shifted from oxidative phosphorylation to glycolysis in the same way as cancer [17].
Sometimes genetic factors confer a predisposition to cellular proliferation and the development of PPA (hereditary PAH). TGF-β signalling generally has a negative effect on cell growth, controlling cell proliferation and apoptosis. The bone morphogenetic type 2 receptor (BMPR2) belongs to the transforming growth factor-beta (TGF-β) superfamily and is expressed in pulmonary vascular smooth muscle and endothelial cells. BMPR2 normally has a protective role, suppressing smooth muscle cell proliferation and normalising endothelial cell apoptosis. Reduced expression or loss of function from mutation of the BMPR2 gene leads to a loss of its growth inhibitory effect and a tendency towards the development of PPA [18]. Genetic predisposition to PPA also occurs less commonly from mutation of downstream mediators (e.g., cytoplasmic signalling proteins- smad proteins) or key regulators (e.g., activin A receptor kinase-1 mutations) in TGF-β signalling [19].
The vascular endothelium, vascular remodelling and inflammatory mechanisms in PAH will be discussed further in Chap. 2.
Pulmonary Veno-occlusive Disease and Pulmonary Capillary Haemangiomatosis
In most conditions associated with PAH the pulmonary veins do not offer a significant contribution to the haemodynamic compromise in the pulmonary circulation and cellular proliferation is not present in the capillary bed. However, this is not the case in pulmonary veno-occlusive disease (PVOD) and pulmonary capillary haemangiomatosis (PCH) which belong to a rare subset of PAH. In PVOD, patients develop significant fibro-occlusive lesions, muscularisation and inflammation of the septal veins and pre-septal venules in addition to a degree of distal pulmonary arteriopathy [20]. In PCH, cellular proliferation arises in the alveolar capillaries. Invasion of adjacent vascular, pulmonary and bronchial structures is typically seen, often associated with diffuse alveolar haemorrhage [21]. Both PCH and PVOD can clinically masquerade as idiopathic PAH but the distinction is extremely important for patient management as pulmonary vasodilators may be deleterious in these patients.
The Pathophysiology of Other Clinical Groups
The development of PPA is reserved to conditions which are classified in the subgroup PAH. Although occlusive lesions and plexiform lesions are exclusive to PPA, endothelial dysfunction leads to pulmonary arterial vasoconstriction, structural remodelling and cellular proliferation in other PH clinical groups. This includes CTEPH [22], PH in the setting of chronic lung disease [23] and left heart disease [24].
Left Heart Disease
Patients classified as having PH secondary to left heart disease (for example left ventricular failure or mitral valve disease) have an elevated left atrial filling pressure which is defined at right heart catheterisation by an elevated pulmonary wedge pressure (PWP >15 mmHg). These patients initially develop a passive rise in mPAP as a consequence of elevated back pressure to the pulmonary circulation from an increase in left atrial filling pressure. This can be associated with a normal or slightly elevated PVR, pulmonary perfusion abnormalities and vascular remodelling. With disease progression, there is superimposed pulmonary vasoconstriction and further vascular remodelling and patients develop established pulmonary vascular disease, associated with significant increases in PVR.
Chronic Lung Disease
Physiological pulmonary vasoconstriction occurs in response to alveolar hypoxia as an important way of matching regional blood flow and alveolar ventilation. In chronic lung disease and or chronic hypoxia, hypoxic vasoconstriction has a key role in PH pathogenesis. However, endothelial dysfunction and inflammation are believed to play the major role in the pathogenesis of PH secondary to chronic lung disease [25].
Chronic Thromboembolic Pulmonary Hypertension
CTEPH is diagnosed when chronically obstructed pulmonary arteries from pulmonary thromboemboli (PE) are associated with pathologically elevated PVR and pulmonary arterial pressure [26]. In this condition, unresolved thrombi become organised and remodel to cause chronic fibrous obstructions within the pulmonary arteries which resists blood flow [27].
The severity of pulmonary artery pressure elevation does not necessarily correlate with the degree of pulmonary vascular obstruction visualised on imaging, nor does the level of PVR necessarily correlate with similar degrees of vascular obstruction associated with acute PE. This is due to progressive distal vasculopathy development in both the occluded and non-occluded regions [28–31]. Muscular hypertrophy resulting in vasoconstriction and fibrointimal proliferation narrows the lumen of the distal pulmonary arteries [22]. It is therefore both the extent of chronic vascular obstruction from organised thrombi and secondary small vessel arteriopathy that contribute to elevated PVR in CTEPH.
CTEPH appears to develop in only 4 % of patients who have previously been diagnosed with acute PE [32], although 60 % of patients that are diagnosed with CTEPH have never had a clinically apparent acute PE [33, 34]. Mechanisms implicated in the pathogenesis of why some patients develop CTEPH include recurrence of PE, in situ thrombosis, unsuccessful resolution of the initial PE or its propagation into branch pulmonary vessels [35]. Abnormalities in the clotting cascade, platelets and endothelium appear to interact in the coagulation process, initiating or exacerbating the development of thromboemboli and/or in situ thrombosis. Some patients have an identifiable abnormality associated with thrombophilia such as protein S or C deficiency or the presence of antiphospholipid antibodies [36].
Pulmonary thromboendarterectomy is the treatment of choice for selected CTEPH patients, offering surgical removal of proximal pulmonary arterial obstruction and therefore potential cure. However, patients with significant distal disease do not benefit from surgery. This will be discussed further in Chap. 4.
Right Ventricular Failure
The pulmonary circulation is normally a low resistance, low pressure network. It can ordinarily accommodate increasing pulmonary blood flow, even during maximal exertion, without a rise in pulmonary arterial pressure. This occurs from passive changes in the compliant pulmonary arteries and capillary beds. Vessels distend and microcirculatory reserves are recruited to lower PVR and to maintain a low pressure circulation [37]. Similarly, early pulmonary vascular disease can be compensated for and is therefore typically subclinical.
The normal low pressure and resistance in the pulmonary circulation offers a low afterload to the right ventricle (RV). It is therefore not adapted to work at high pressure but is adapted to accommodate the variable haemodynamic demands placed upon it. The RV is relatively thin walled and compliant compared with the thick walled left ventricle, but it hypertrophies as the pulmonary artery pressure increases. Initially the RV can overcome the increased afterload associated with a higher PVR [38] but later, cardiomyocyte contractility declines and as the contractile force weakens the RV dilates. RV dilatation increases wall tension and oxygen demand whilst decreasing perfusion. This further decreases contractile function and a vicious cycle ensues [39]. The contractile reserve and the transition from RV hypertrophy to failure is dependent not only on the severity and time course of increasing afterload but also on the degree of oxidative stress, reduced myocardial perfusion, neuro-hormonal and immune activation and the response of the RV to this including remodelling and fibrosis [37, 38, 40–43].
Clinical Presentation
PH typically has an insidious onset, with non-specific symptoms. Clinical findings are commonly attributed to concomitant diseases. It is therefore a challenging diagnosis to consider and investigate, especially early in the disease process. Failure to diagnose or incorrect diagnosis is common [45]. A reported median of 14 months from onset of symptoms to diagnosis has been found [46]. A high clinical index of suspicion is necessary, especially in patient groups at increased risk of developing PH.
Symptoms
Patients with early PH may be asymptomatic. The first symptoms that usually develop are fatigue and dyspnoea on exertion. With disease progression symptoms become more apparent, as the PVR escalates and cardiac output falls. Breathlessness with diminishing exertion and then at rest ensues. Palpitations, chest pain (right ventricular angina), presyncopal episodes and later syncope can develop. As the right heart fails, symptoms advance to include peripheral oedema and abdominal swelling from ascites. Eventually, without successful treatment, patients die from RV failure [38, 47, 48].
Functional Classification
Patients with PH are classified according to the World Health Organisation (WHO) 1998 New York Heart Association functional classification (FC). This classifies the patient according to the symptoms they experience during daily living (Table 1.1) [1]. A patient’s functional class is important in determining their prognosis and helps determine disease severity and response to treatment.
Table 1.1
Functional classification of patients with pulmonary hypertension according to the World Health Organisation, modified after the New York Heart Association functional classification [1]
Class | Description |
|---|---|
I | Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnoea or fatigue, chest pain, or near syncope. |
II | Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnoea or fatigue, chest pain, or near syncope. |
III | Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnoea or fatigue, chest pain, or near syncope. |
IV | Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnoea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity. |
Physical Examination
Abnormal physical findings may be subtle in patients with PH and can be missed. A loud pulmonary component to the second heart sound may be heard from forceful pulmonary valve closure secondary to elevated pulmonary artery pressure. The jugular venous pressure may depict a prominent ‘a’ wave from high RV filling pressures. With RV dilatation the tricuspid apparatus can dilate resulting in functional regurgitation and accentuated ‘v’ waves identifiable in the jugular venous waveform. Equally the pansystolic murmur of tricuspid regurgitation may be audible. A left parasternal lift can occur as a consequence of RV hypertrophy. A right ventricular fourth heart sound (immediately prior to the first heart sound) may result from abnormal turbulent blood flow as the right atrium contracts and forces blood flow into a stiffened RV. Alternatively with RV dysfunction and volume overload a third heart sound may follow the second heart sound in mid diastole during ventricular filling. With RV failure, jugular venous distension, hepatomegaly, ascites and peripheral oedema are common features. Arrhythmia may develop as a consequence of atrial dilatation or RV dilatation, hypertrophy or fibrosis and rhythm disturbance is important to recognise on clinical examination as it can impede cardiac function.
Diagnosis
PH is defined by the resting mPAP measured at right heart catheterisation (> = 25 mmHg). Therefore, the diagnosis of PH ordinarily requires invasive confirmation.
The normal value of mPAP is 14 ± 3 mmHg [1, 49]. Patients with borderline mPAP values (17–24 mmHg) should be prospectively monitored for the development of PH and research into the significance and suitable management of these patients is ongoing.
Right Heart Catheterisation
Right heart catheterisation (Fig. 1.2) measures the pressure, flow and resistance in the cardiopulmonary circulation, including measurement of the downstream left atrial pressure (PWP). Cardiac shunting can also be identified and measured by oxygen saturation assessment [50].
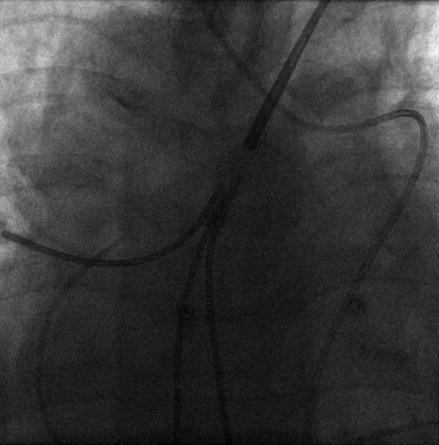
Figure 1.2
Plain radiograph showing a right heart catheter in the pulmonary artery of a patient with dextrocardia (precordial leads also shown)
PWP is determined by wedging the tip of the right heart catheter with the balloon inflated in the pulmonary artery. The forward flow of blood is temporarily stopped within the small vessel where the catheter lies. When there is no venous obstruction, the measured pressure is the back pressure transmitted from the left atrium, as there are no anatomical valves in the pulmonary veins [51, 52]. A PWP of greater than 15 mmHg defines a significant contribution to raised mPAP from pressure beyond the pulmonary capillary bed, for example from left atrial hypertension.
In general the PWP is not greater than 15 mmHg in patients with PAH [53]. However, the size, shape and compliance of one ventricle has a direct effect on the other ventricle through their mechanical interaction, including a shared septum, myofibres and a joint pericardial space encased by a fibrous pericardium. Therefore, left ventricular dysfunction can result from significant RV dysfunction, and this can lead to elevated left atrial filling pressure and PWP in PAH [54]. PWP may also be elevated (or normal) in pulmonary veno-occlusive disease. This depends on the size of the veins involved and the degree of collateral communication between the affected venous beds [20].
Ohm’s law pertaining to electrical resistance states that resistance equals pressure divided by flow. Therefore, PVR is calculated by dividing the transpulmonary pressure gradient (mPAP – PWP (mmHg)) by cardiac output (L/min). This is an important measurement determined by right heart catheterisation which is commonly measured in arbitrary units called Wood Units (WU). One Wood Unit is 1 mmHg per litre per minute or 80 dynes.sec.cm−5 [55]. PVR is abnormal when above 2WU but is defined as significantly raised when greater than 3WU [53, 56, 57]. A pathological elevation in PVR signifies the presence of significant pulmonary vasculopathy. PVR greater than 3WU is included in the definition of PAH [53].
Right heart catheterisation identifies important prognostic variables for patients with PH. Mean right atrial pressure, cardiac index (cardiac output adjusted for body surface area) and PVR have been shown to predict mortality in national PAH registries [58–60]. Notably PVR is a more important variable than mPAP in the assessment of the severity and prognosis of PH [61]. Progressive pulmonary vasculopathy is translated into an increasing PVR. Paradoxically, with increasing afterload the RV starts to fail and the cardiac output reduces, and this can result in a fall of mPAP despite advancing disease.
Right heart catheterisation procedures are generally well tolerated [62] but due to their invasive nature significant serious adverse events (1.1 %) and mortality (0.1 %) have been reported in multicentre large patient series [63]. However, right heart catheterisation accurately defines an individual’s pulmonary haemodynamics to establish a diagnosis of PH. It aids treatment decisions including suggesting contributing factors, identifying important prognostic variables and assessing disease progression.
Non invasive Investigations
Although right heart catheterisation is the gold standard diagnostic investigation for PH, non invasive estimates are advantageous in terms of cost, availability and risk. They are used to help identify those patients that require further investigation into a diagnosis of PH but they cannot reliably exclude the diagnosis. Non invasive investigations are important in establishing conditions causing or contributing to the diagnosis of PH such as thromboemboli, left heart or lung disease.
Echocardiography
Echocardiography is used to aid the diagnosis of PH initially, risk-stratify patients and monitor their progress. It is also important in the identification of valvular pathology, cardiac shunts (especially when contrast is used) and primary myocardial disease, that may contribute to a diagnosis of PH [64].
Transthoracic echocardiography determines the pulmonary arterial pressure to aid the identification and monitoring of PH patients. The most common method is determining the pulmonary artery systolic pressure by the velocity of measurable tricuspid regurgitation (Fig. 1.3). A meta-analysis of this method revealed a moderate diagnostic accuracy of 83 % sensitivity (95 % confidence interval 73–90 %) and 72 % specificity (95 % confidence interval 53–85 %) when compared to invasive measurement [65]. The wide confidence intervals highlight the weakness of echocardiography as a sole investigative or screening tool in PH.
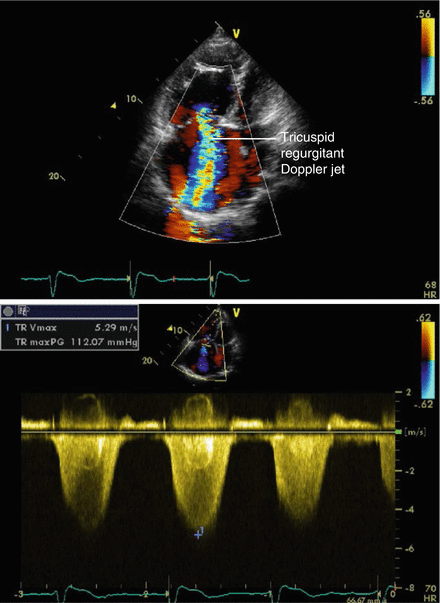
Figure 1.3
Pulmonary artery systolic pressure estimation by the continuous wave Doppler tracing of the tricuspid regurgitant jet velocity in a patient with pulmonary hypertension. From a tricuspid regurgitant velocity of 5.29 m/s the right ventricular systolic pressure can be calculated to 135–140 mmHg, equivalent to pulmonary artery systolic pressure in the absence of right ventricular outflow obstruction
Increasing afterload in PH stresses the thin walled and distensible right ventricle to forge its shape, size and functional state. When adequate echocardiography views are obtained, the morphology and function of the RV can be assessed to serially evaluate RV dysfunction and suggest the presence of PH. In addition, the discussed interaction between the right and left ventricle can be visualised, and the size and pressure within the right atrium can be reviewed. The presence of a pericardial effusion can be established easily by echocardiography which has been shown to predict death or transplantation in PAH [66, 67].
As PH cannot reliably be defined by a cut off pulmonary artery pressure value using echocardiography alone, current guidelines suggest incorporating other echocardiography measures and determining the probability of PH. PH is unlikely in the presence of pulmonary artery systolic pressures below 36 mmHg in the absence of additional echocardiogram variables suggestive of PH, possible at higher values and likely when greater than 50 mmHg (Table 1.2) [1, 49]. It is essential that the full diagnostic potential of echocardiography is used when assessing PH.
Table 1.2
Arbitrary criteria for estimating the presence of pulmonary hypertension (PH) by echocardiography, according to international guidelines [1]
Echocardiography diagnosis of PH | Tricuspid regurgitant peak velocity | Pulmonary artery systolic pressure* | Additional echocardiogram variables suggestive of PH |
|---|---|---|---|
Unlikely | ≤2.8 m/s | ≤36 mmHg | No |
Possible | ≤2.8 m/s | ≤36 mmHg | Yes |
Possible | 2.9–3.4 m/s | 37–50 mmHg | Yes/no |
Likely | >3.4 m/s | >50 mmHg | Yes/no |
Other Routine Investigations
An electrocardiogram is important in PH to identify and diagnose arrhythmia so that suitable management can be initiated. The electrocardiogram may also show evidence of right atrial enlargement, right ventricular strain or hypertrophy [68] to suggest a diagnosis of PH. However, it is not a sufficient screening tool for the presence of PH, especially in early disease [47].
At present, the place for radiological investigation in patients with suspected PH is mainly in the identification of lung diseases or CTEPH. However, recognition of pulmonary artery enlargement or dilatation of cardiac chambers from imaging can suggest the diagnosis of PH (Fig. 1.4).
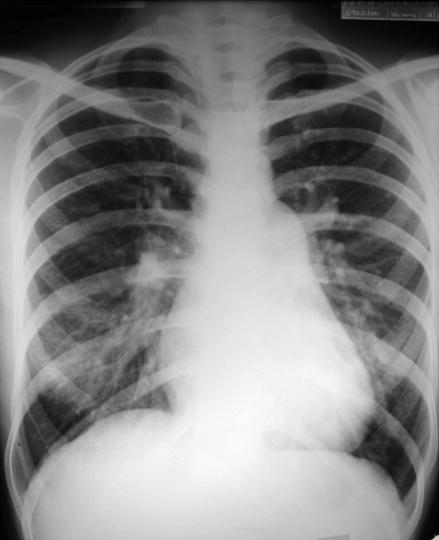
Figure 1.4
Chest radiograph showing plethoric pulmonary arterial vasculature with right ventricular enlargement elevating the cardiac apex and reduced size of the aortic knuckle in a patient with pulmonary hypertension from atrial septal defect
A baseline chest radiograph is considered routine and may reveal features suggestive of PH, left ventricular impairment (Table 1.3), or lung disease.
Table 1.3
Chest radiograph features suggestive of left ventricular impairment
Chest radiograph feature | Stage of left ventricular impairment progression |
|---|---|
Cardiac enlargement | Variable, widely prevalent in chronic cases |
Upper lobe venous redistribution | Elevated pulmonary venous pressure |
Central venous prominence | Elevated pulmonary venous pressure |
Indistinctness of the central perihilar vasculature | Interstitial oedema |
Thickening of the minor fissure | Interstitial oedema |
Pleural effusion | Interstitial oedema |
Diffuse air space disease | Alveolar oedema |
Computed tomography (CT) offers a simultaneous assessment of the pulmonary vasculature (pulmonary angiography) and lung parenchyma (high resolution CT) and is the main diagnostic strategy in PH radiological investigation (Fig. 1.5) [69, 70]. The presence of acute or chronic thromboembolic disease related to CTEPH or lung diseases such as emphysema can be recognised by CT. CT imaging can also be helpful in identifying PVOD and PCH which are typically challenging to diagnose. PVOD typically demonstrates features of pulmonary interstitial oedema without engorgement of the main pulmonary veins or left sided chambers (as the occlusion is in the more proximal post capillary venules). Comparatively, extensive ground glass nodules are typical in PCH [71].
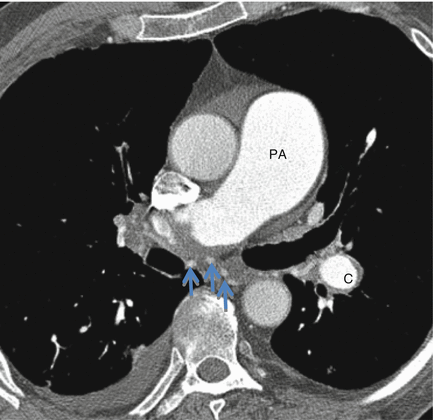
Figure 1.5
CT pulmonary angiogram showing bronchial artery hypertrophy (arrows) in a patient with chronic thromboembolic pulmonary hypertension. Note the marked enlargement of the pulmonary artery (PA) and the rim of clot around the left lower lobe pulmonary artery (C)
Ventilation perfusion scintigraphy is used to further investigate the presence of CTEPH by identifying the mismatch between perfusion and ventilation from vessel occlusion [72, 73].
Pulmonary function testing is performed to identify lung disease as a contributor to PH. With adequate technique, physiological impairment from underlying respiratory disease can be identified and quantified. However, even without concomitant lung disease, a reduction in carbon monoxide diffusion is typically demonstrated in PAH. Damage to the pulmonary vascular bed in PAH results in decreased capillary blood volume and flow and a reduction in the surface area available for gas diffusion [74, 75]. An unexpected reduction in a patient’s gas diffusion should guide the physician to consider a diagnosis of PAH.
Routine haematological and biochemical investigations in PH include full blood count, urea and electrolytes and liver function testing. Liver function testing is important to recognise a predisposition to and potential cause of PH, with consideration given to liver ultrasound. Similarly, autoimmune profiles may help identify underlying connective tissue diseases, and HIV testing in patients with PH is recommended [1]. Screening for thrombophilia or sickle cell disease is appropriate in patients with a suggestive history. Genetic testing is important in idiopathic and heritable PAH including the offer of genetic counselling and testing of relatives.
Brain natriuretic peptide (BNP) and the biologically inactive N-terminal segment (NT-proBNP) can be measured in the blood and are released from the myocardium in response to wall stress. In the presence of right ventricular dysfunction these markers typically rise and therefore they are useful for monitoring disease progression in PH. An elevated or increasing level of BNP or NT-proBNP is a poor prognostic sign suggesting progressive right ventricular dysfunction [76]. However, in early disease before significant cardiac dysfunction, their levels can be normal and their levels will also rise in response to left ventricular dysfunction. An elevation in BNP or NT-pro BNP level is therefore not specific to a diagnosis of PH nor does a normal level exclude the condition [77, 78].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


