Fig. 7.1
Interaction between the sympathetic nervous system, the adrenal gland, and the cardiovascular system in chronic heart failure. Under physiological conditions, sympathetic nerves innervating the heart release noradrenaline to modulate cardiac function via α1– and β-adrenoceptors. Adrenaline reaching the heart as a hormone is secreted from the adrenal gland. α2-adrenoceptors operate as inhibitory feedback receptors to control maximal noradrenaline and adrenaline secretion. In heart failure, feedback loops activate the central sympathetic system to increase the sympathetic outflow to the heart
The Autonomic Nervous System
In general, the peripheral autonomic nervous system can be divided into several groups: (1) the sympathetic nervous system, (2) the parasympathetic nervous system, and (3) the enteric nervous system [10]. In addition to efferent nerves, the cardiovascular system is also innervated by sensory neurons.
Sympathetic Nervous System
Sympathetic pathways are composed of two types of neurons, termed pre- and postganglionic sympathetic neurons . The bodies of primary, preganglionic sympathetic neurons are located in the intermediolateral column of the thoracic and lumbar regions of the spinal cord. Their axons leave the central nervous system (CNS) to enter the paravertebral chains on both sides of the vertebral column s10]. In sympathetic ganglia within the paravertebral chains, primary sympathetic neurons connect with the secondary, postganglionic sympathetic neurons. In addition to the paravertebral ganglia, postganglionic sympathetic neurons are also located in prevertebral ganglia which are located in close vicinity of the aorta and its main abdominal branches [10] .
Postganglionic sympathetic neurons innervate most tissues and organs of the body as a dense plexus of sympathetic axons. In contrast to many neurons in the CNS, sympathetic axons do not connect with their target cells as nerve terminals at a single synapse. Sympathetic (as well as parasympathetic) axons have several branches and neurotransmitter vesicles are stored in multiple varicosities along the axon to form a “pearl and chain” architecture ([11]; Fig. 7.1). Sympathetic varicosities and target cells form a specialized neuromuscular junction at the ultrastructural level [12].
The adrenal gland is an essential part of the sympathetic system, as chromaffin cells in the medulla resemble postganglionic sympathetic neurons lacking an axon [13]. Sympathetic neurons synthesize and release noradrenaline (norepinephrine) as their main neurotransmitter (Fig. 7.2). Chromaffin cells express an additional enzyme, phenylethanolamine N-methyltransferase (PNMT), which converts 90 % of the synthesized noradrenaline into adrenaline ([13]; Fig. 7.2). Thus, adrenaline reaches its target cells and organs as a hormone via the circulation whereas noradrenaline derives from sympathetic nerves and also—to a smaller degree—from the adrenal gland .
Pre- and postganglionic sympathetic neurons contain additional co-transmitters, mostly neuropeptides as well as adenosine triphosphate (ATP) [13]. Immunohistochemical studies have revealed that sympathetic pathways which innervate particular tissues or organs contain distinct combinations of co-transmitters [13]. This “chemical coding” provides a cellular basis for specific activation of discrete tissues and organs by specific sympathetic pathways and fibers [14]. In the heart, sympathetic activation increases heart rate (chronotropic effect), conduction velocity (dromotropic), speed of contraction (inotropic), and relaxation (lusitropic). In the vascular system, sympathetic activation is linked with both vasoconstriction as well as vasodilatation to control blood pressure , peripheral resistance, and organ perfusion .
The molecular components involved in catecholamine synthesis, storage, release, and action have been unraveled in detail ([15, 16]; Fig. 7.2). After sequential synthesis of noradrenaline (in sympathetic neurons) or adrenaline in (adrenal chromaffin) cells, catecholamines are stored in neurotransmitter vesicles. In postganglionic sympathetic fibers, neurotransmitter vesicles are accumulated in varicosities, from which the transmitter is secreted upon arrival of an action potential. Once noradrenaline has reached the extracellular space of the heart, it may activate surrounding adrenoceptors (Fig. 7.2).
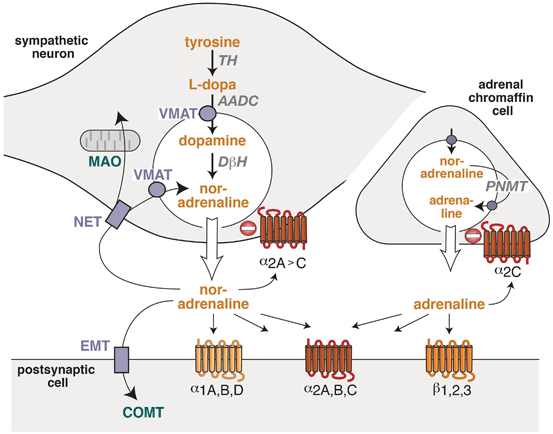
Fig. 7.2
Functional organization of the adrenergic system in sympathetic nerves and in the adrenal gland. In sympathetic nerves, noradrenaline is synthesized from the precursors tyrosine, L-dopa, dopamine and it is stored in synaptic vesicles. In the adrenal gland, noradrenaline is converted into adrenaline . Both catecholamines activate different G-protein-coupled receptors which are located in the post- or presynaptic membranes. Pre- and postsynaptic transporters mediate cellular or vesicular uptake of catecholamine (symbols in light blue), and enzymes (green) metabolize catecholamines. α 1A,B,D’ α 2A,B,C’ β 1,2,3 adrenoceptor subtypes, AADC aromatic amino acid decarboxylase, COMT catechol-O-methyltransferase, DβH dopamine β-hydroxylase, EMT extraneuronal catecholamine transporter, MAO monoamine oxidase, NET noradrenaline transporter, TH tyrosine hydroxylase, VMAT vesicular monoamine transporter, PNMT phenylethanolamine N-methyltransferase
The cellular functions of adrenaline and noradrenaline are mediated by G-protein-coupled receptors which are termed “adrenoceptors” and are located in the plasma membrane of many different cell types. Based on their homology, physiological function, and pharmacological profiles, these adrenoceptors were grouped into α- and β-adrenoceptor subtypes [17]. Until now, nine adrenoceptors have been identified by molecular cloning: three α1-adrenoceptors (α1A, α1B, α1D), three α2-adrenoceptors (α2A, α2B, α2C), and three β-adrenoceptors (β1, β2, β3) [17]. For the elucidation of the structure, function, and regulation of adrenoceptors, the Nobel prize in chemistry was awarded to Dr. Robert J. Lefkowitz and Dr. Brian Kobilka in 2012 [18, 19] .
Adrenoceptors have been identified in postsynaptic membranes as well as in the presynaptic membrane of sympathetic (and many other) neurons [20]. Presynaptic receptors may be separated into two groups, depending on whether they are activated by their neuron’s own transmitter (“autoreceptors”) or by other neurotransmitters or signals derived from other cells (“heteroreceptors”) [16]. Among the sympathetic autoreceptors, α2-adrenoceptors are inhibitory receptors while β-adrenoceptors stimulate noradrenaline release. Presynaptic α2-adrenoceptors are important components of a regulatory feedback which limits the maximal amount of noradrenaline release (Fig. 7.2). In sympathetic neurons, α2-adrenoceptors are activated by extracellular noradrenaline and inhibit further transmitter exocytosis [20]. Mouse models lacking individual α2-adrenoceptor subtypes have been important tools to identify the receptor subtype and its function within this feedback loop. In sympathetic neurons, α2A– and α2C-receptors function together as presynaptic inhibitors ([21]; Fig. 7.2). Both receptor subtypes inhibited neurotransmitter release in several neuron types [22–24] and differed in their functional properties: α2A-adrenoceptors inhibited noradrenaline secretion at higher action potential frequencies than α2C-receptors [21]. This finding was consistent with longer persisting activation of intracellular signaling after agonist removal for α2C-receptors than for α2A-receptors [25]. Thus, due to a higher affinity for noradrenaline , α2C-adrenoceptors were suggested to act as feedback inhibitors at low levels of sympathetic activity and α2A-receptors as inhibitors at higher or maximal levels of sympathetic activity [21]. A similar feedback control mechanism has also been described for the adrenal gland (see Sect. 5 below) .
Parasympathetic Nervous System
Similar to the sympathetic system , also the parasympathetic part is composed of two neuron types which are connected to each other in series [10]. However, in contrast to the sympathetic system, parasympathetic ganglia are located close to or even within its target organs. Preganglionic parasympathetic neurons responsible for cardiac control originate in the dorsal motor nucleus and the nucleus ambigous and leave the CNS via the vagal nerve. In cardiac ganglia, preganglionic neurons innervate the postganglionic parasympathetic neurons. Axons from these cells innervate several regions of the heart with particular dense levels of innervation in sinus and atrioventricular nodes and in the atria [10]. The major neurotransmitter of the parasympathetic system is acetylcholine which may be released together with additional neuropeptides and ATP as co-transmitters [10]. Some parasympathetic nerves synthesize nitric oxide which may act like a neurotransmitter, although it is not stored in neurons . All ganglionic transmissions in both branches of the autonomic nervous system are mediated by acetylcholine which activates nicotinic receptors of postganglionic neurons. Recent histological studies indicate that not only atria but also cardiac ventricles are innervated by parasympathetic nerves. In particular, the ventricular endocardium and epicardium contain fine networks of parasympathetic fibers [26]. Parasympathetic activation decreases heart rate and atrioventricular conduction with minimal or no effects on cardiac contractility (for discussion, see [27]) .
Sympathetic and parasympathetic systems interact functionally by their opposing effects on postsynaptic cells of the cardiovascular system . In addition, they also interact on the presynaptic side by presynaptic receptors ( “heteroreceptors”) which modulate the neurotransmitter release of the other branch of the autonomic system [16]. Thus, noradrenaline alters parasympathetic acetylcholine release and acetylcholine acts at sympathetic nerves to control noradrenaline secretion.
Development of the Cardiac Sympathetic Nervous System
The heart is not homogeneously innervated by autonomic nerves and the development of the pattern of innervation during the prenatal period has been studies in several species [28]. The highest density of sympathetic nerves has been observed in the subepicardium and in the central conduction system [29–31]. All autonomic nerves which innervate the heart originate from neural crest cells during embryonic development [28]. The neural crest not only is the source for sympathetic and parasympathetic neurons but also gives rise to cardiac sensory neurons. In the adult organism, these cardiac sensory neurons are located in the dorsal root ganglia and are part of afferent cardiovascular reflex pathways. They also mediate pain sensation in response to cardiac ischemia [32].
During embryonic development, growth and migration of autonomic nerves from the neural crest to the heart and other target tissues are controlled by a number of neurotrophic factors [28]. Nerve growth factor (NGF) has been identified as one of the key factors for sympathetic growth during development and disease (Fig. 7.3). The final degree of sympathetic innervation of the heart correlates with the level of expression of NGF in the target tissue. During development, NGF is expressed in several regions of the CNS, but mature cardiomyocytes also express high levels of NGF protein [33, 34]. This finding was further supported by studies in mice lacking or overexpressing NGF or its receptor TrkA (tropomyosin related receptor kinase). Ablation of NGF expression as well as deletion of the TrkA gene in mice led to severe impairment of sympathetic ganglion development soon after birth [35, 36]. In contrast, overexpression of NGF under control of a cardiac myocyte-specific promoter led to sympathetic hyperinnervation and increased tissue norepinephrine levels in the heart [37]. NGF plays a similar trophic role during the development of the cardiac sensory system. Thus, cardiac NGF is essential to prevent apoptosis of sympathetic neurons and to guide innervation of the heart and other sympathetic target tissues and organs ([38]; Fig. 7.3).
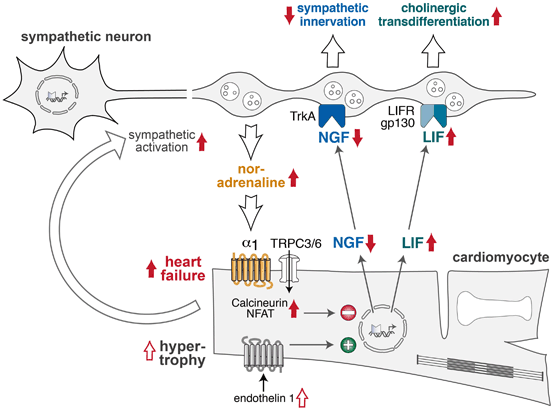
Fig. 7.3
Interactions between sympathetic nerves and cardiomyocytes in cardiac hypertrophy and failure. During development, endothelin-1 activates transcription and release of nerve growth factor (NGF) from cardiomyocytes . NGF activates neuronal TrkA receptors to facilitate sympathetic neuron outgrowth and thus determines sympathetic innervation density of the heart. In the failing heart, increased noradrenaline and Ca2 + influx through transient receptor-operated channels (TRPC3/6) activate cardiomyocyte calcineurin/NFAT signaling to inhibit NGF expression and reduce sympathetic innervation. Enhanced LIF expression in the failing heart stimulates sympathetic neurons to express cholinergic genes (“cholinergic transdifferentiation”). LIF leukemia inhibitory factor, LIFR/gp130 LIF receptors, NFAT nuclear factor of activated T cells, TrkA tropomyosin-related kinase receptor
Several factors which control cardiac expression of NGF have been identified [28]. Endothelin-1 (ET-1) and noradrenaline play opposing roles in the regulation of cardiomyocyte NGF expression (Fig. 7.3). ET-1 induces NGF expression in isolated cardiomyocytes in vitro and in the heart in vitro [33]. In ET-1-deficient mice, cardiac NGF expression and sympathetic innervation of the heart were significantly diminished [33]. Stellate ganglia from ET-1-deficient mice showed loss of sympathetic neurons due to apoptosis and these phenotypes could be rescued by cardiac-specific overexpression of NGF [33]. Mechanical stretch and activation of α1-adrenoceptors by noradrenaline were identified as repressive factors for cardiac NGF [39]. Downstream of α1-adrenoceptors, calcineurin/nuclear factor of activated T cells (NFAT) activation decreased cardiomyocyte expression of NGF [39]. Semaphorin 3a was identified as a second factor which inhibits cardiac sympathetic innervation during the development [40].
In contrast to the sympathetic system, less information exists about the prenatal development of cardiac parasympathetic nerves. Glial cell line-derived neurotrophic factor (GDNF) plays an important neurotrophic role in guiding parasympathetic innervation of the mouse heart via its receptors GFRα2 and Ret [41].
Sympathetic Nerves in Heart Disease
In chronic heart failure, the structure and function of the sympathetic system are altered. Reduced cardiac performance leads to a long-term, persistent sympathetic activation. Clinically, tachycardia has long been known as a symptom of heart failure, reflecting increased stimulation of the sinus node by noradrenaline released from cardiac sympathetic nerves [42]. Patients with heart failure showed elevated circulating and urinary levels of catecholamines [3, 43]. At the same time, the myocardial noradrenaline content was lower in failing hearts as compared with non-failing human hearts [43, 44]. These findings are consistent in both structural and functional alterations of the cardiac sympathetic system in chronic heart failure.
Functional Alterations in Heart Failure
In human and experimental heart failures, sympathetic outflow to the heart, kidney, and vasculature are increased ([27]; Fig. 7.1) . Functionally, increased sympathetic activity aims to maintain blood pressure and cardiac output by stimulating cardiac contractility and inducing vasoconstriction. On the venous side, vasoconstriction enhances cardiac preload and on the arterial side it elevates peripheral resistance thus supporting blood pressure . By its action on renal proximal tubular cells, noradrenaline may, similar to angiotensin II, facilitate sodium and water retention. A comprehensive analysis of cardiac noradrenaline kinetics in normal subjects and in patients with heart failure identified the processes which are altered in heart failure [45]. Increased adrenergic drive to the failing heart resulted in increased cardiac release of noradrenaline and was accompanied by decreased efficiency of the presynaptic uptake mechanisms (Fig. 7.3). Thus, spillover of noradrenaline from the cardiac interstitial space into the blood stream was significantly enhanced in patients with heart failure [45]. Remarkably, elevated circulating concentrations of noradrenaline correlated with increased mortality in heart failure [3].
Chronic exposure to high levels of noradrenaline and enhanced β-adrenergic signaling are toxic to the heart [46–48] . On the postsynaptic side, chronic adrenergic activation leads to cardiomyocyte hypertrophy and interstitial fibrosis and several other changes which are summarized by the term “cardiac remodeling” [6, 49]. Several molecular events are combined to desensitize cardiac β-adrenergic signaling in response to chronic stimulation [19, 50]. Desensitization occurs at several levels, including downregulation of β1-adrenoceptors in cardiomyocytes , uncoupling of β1– and β2-receptors from their G proteins by phosphorylation of receptors and arrestin binding, and by redistribution of β2-receptors in the plasmamembrane [51, 52]. Altogether, these changes lead to a decrease in the inotropic reserve of the heart after chronic adrenergic stimulation .
Heart Failure with Preserved Ejection Fraction
Half of all patients with chronic heart failure have a preserved left ventricular ejection fraction (HF-pEF), but prognosis of these patients may be similar to patients with failure and reduced ejection fraction. Several cardiac and extracardiac factors have been suggested as key factors in the pathogenesis of the disease [55, 56]. However, only limited data are available concerning the activity of neurohumoral systems in HF-pEF. In a small study, patients with HF-pEF had significantly elevated plasma noradrenaline levels compared with control subjects [57]. However, additional studies suggest that stable patients with HF-pEF have lover levels of sympathetic activation than patients with reduced left ventricular function. HF-pEF patients may show similar rises in sympathetic activity during phases of acute decompensation. Thus, the role of neuroendocrine activation in heart failure with preserved ejection fraction remains unclear at present [56].
Alterations in Afferent Autonomic Reflexes and in the CNS
The mechanisms and pathways linking cardiac dysfunction with activation of central autonomic neuronal pathways have been studied intensively (for recent reviews, see [58, 59]). The prevailing model suggests that sympathetic activation is initiated by left ventricular systolic dysfunction which activates cardiovascular baroreceptors. Central reflex pathways which are activated in response to the baroreflex elicit a generalized increased in central sympathetic outflow [58]. However, impaired cardiac performance is by itself not sufficient to account for the elevated levels of sympathetic activation. A spectrum of afferent nerves relays signals from the heart, lung, and vascular system to the CNS. The precise mechanisms of sympathetic augmentation which increase adrenergic activity to higher levels and longer periods than required for maintenance of cardiovascular function are only partially understood [58]. Indeed, several lines of evidence suggest a more selective rather than generalized autonomic activation in heart failure. In early cardiac dysfunction, selective enhancement of cardiac noradrenaline release which precedes the elevation of total body noradrenaline spillover which is characteristic for advanced-stage heart failure [60]. Arterial and cardiopulmonary baroreflexes were found to be altered in human heart failure , thus contributing to enhanced sympathetic stimulation (for review, see [59]). Several alterations in the central circuitry involved in these reflexes, including enhanced angiotensin II signaling in circumventricular organs, have been uncovered [61, 62].
Structural Changes of Sympathetic Nerves
Cardiac disease alters sympathetic innervation of the heart [31, 63–65]. After myocardial infarction, sympathetic nerves degenerate within and distal to the site of ischemia but may regenerate after infarct healing. However, reinnervation of the heart after injury may be heterogeneous and has been suggested to play a key role in arrhythmia development [66]. In failing hearts, sympathetic innervation decreases leading to depletion of cardiac noradrenaline content [67]. In diabetes mellitus, cardiac NGF expression was found to be reduced and was associated with a decreased sensory innervation of the heart [68, 69].
In addition to its central role during the development of cardiac sympathetic innervation, NGF may also be important for structural alterations of the sympathetic system in the failing heart ([70–72]; Fig. 7.3). In experimental hypertrophy and heart failure induced by chronic noradrenaline infusion, cardiac expression of NGF was suppressed by the activation of calcineurin/NFAT signaling due to elevated intracellular Ca2+ (Fig. 7.3). Activation of transient receptor potential channels TRPC3/6 was shown to be important for initiating this pathway [39, 73, 74]. As a consequence of NGF repression, cardiac sympathetic innervation density was significantly reduced (Fig. 7.3). In a rat model of heart failure, the lack of NGF could be rescued by injection of NGF into stellate ganglia thus leading to improved cardiac noradrenaline reuptake and improved cardiac performance [75] .
Several experimental data suggest that sympathetic neurons may alter their phenotype in chronic heart failure. In failing hearts, sympathetic ganglia express genes which are typical for cholinergic or immature neurons [76]. Among the cytokines which are elevated in heart failure, leukemia inhibitory factor (LIF) and cardiotrophin-1 led to transdifferentiation of sympathetic neurons to acquire cholinergic activity via activation of gp130 signaling ([77]; Fig. 7.3). Kimura et al. called this phenomenon “functional denervation due to rejuvenation” [28]. At present, it remains unclear whether transdifferentiation of sympathetic nerves to a parasympathetic phenotype is adaptive or maladaptive in heart failure. Mice lacking gp130 signaling in sympathetic neurons did not show signs of transdifferentiation and had a higher mortality rate in a hypoxia-induced heart failure model than wild-type mice [77]. This finding indicates that transdifferentiation of sympathetic neurons may be protective.
Adrenal Gland in Physiology and Heart Failure
Similar to sympathetic neurons, α2C-adrenoceptors operate as feedback regulators of catecholamine release from adrenal chromaffin cells ([50, 78]; Figs. 7.1 and 7.2). Interestingly, basal feedback control of catecholamine release from adrenal medulla or sympathetic nerves was mediated by different α2-adrenoceptor subtypes [50] . While mice lacking α2A-receptors showed enhanced plasma noradrenaline levels, α2C-deficient mice displayed twofold elevated circulating adrenaline concentrations [50]. At present, it remains unclear which α2-adrenoceptor subtype(s) are essential for adrenal feedback control in different species. While α2C predominates in mouse chromaffin cells [50], α2A is the major subtype in rat adrenal medulla [78]. Both receptor subtypes have also been identified in the human adrenal [79].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


