Fig. 11.1
Human Igf1 gene alternative splicing. Different leader sequences of the Igf1 gene result in two different classes of mRNA variants; class 1 transcripts use exon 1 as leader exon, whereas class 2 transcripts have the leader sequences on exon 2. All possible combinations between signal peptide sequences and terminal exon (5 or 6) can occur in different IGF-I mRNA isoforms. The mature IGF-I peptide is coded by exons 3 and 4. It is a common part of the IGF-I precursor polypeptides and it is derived from post-translational processing of each of the multiple IGF-I precursors, by which the signal peptides and the E-peptides (Ea, Eb, Ec) are removed. The different E-peptides are encoded by three mRNA variants produced by alternative splicing of the 3′ end of the pre-IGF-I mRNA. The first 16 amino acids of the amino-terminal portion of each of the IGF-I E peptides are coded by exon 4, while exons 5 and 6 encode distinct portions of the E-peptides with alternative carboxy-terminal sequences
11.2 The IGF-I Bioregulation System
The IGF-I system consists of IGF-I, which produces three isoforms in humans, the type 1 (IGF-IR) and type 2 (IGF-IIR) IGF receptors, insulin receptor (IR), and several atypical receptors, such as the hybrid IGF-IR/IR [19], as well as of the IGF binding proteins (IGFBPs) [20, 21].
11.2.1 IGF-I Alternative Splicing/Isoforms
The Igf1 gene is a highly conserved sequence in mammals and primates. It contains six exons and as a result of its alternative splicing, different IGF-I mRNA transcripts are produced, encoding for several IGF-I precursor proteins [4]. Specifically, class 1 or class 2 mRNA transcripts are produced by differential splicing of exons 1 or 2, respectively, to the common exon 3. Alternative splicing of exon 5 results also in different mRNA variants containing exon 5, (class B, or IGF-IEb variant), or containing exon 6 (and excluding exon 5) defined as class A (or IGF-IEa variant). A third transcript variant, the IGF-IEc, which corresponds to IGF-IEb in rodents, is also generated by alternative splicing in the human Igf1 gene and contains both exon 5 and 6. All possible combinations between leader exon (1 or 2) and terminal exon (5 or 6) can occur in different IGF-I transcripts [13]. Thus, the corresponding IGF-I protein isoforms, namely the IGF-IEa, IGF-IEb and IGF-IEc in humans, differ by the structure of their E-peptides, on the carboxy-terminal end, and by the length of their amino-terminal signal peptides, however they share the same mature peptide [13, 14], (Fig. 11.1).
Recent studies have shown that the IGF-I splice variants are differentially transcribed in response to varying conditions and pathologies, such as exercise-induced muscle damage [22], myocardial infarction [17, 18], endometriosis [23], and cancer [24, 25]. The differential expression/regulation of the IGF-I splice variants in various pathologies could indicate distinct biological roles of the different IGF-I isoforms, however their particular functions remain, as yet, unclear.
11.2.2 IGF Binding Proteins
IGF-I is constitutively produced from many tissues and can also be released into the circulation [19]. In contrast with other growth factors, IGF-I (and IGF-II) can associate with specific IGF binding proteins (IGFBPs) in plasma and tissues [3]. At least six IGFBPs modulate the biological actions of IGF-I, as they bind IGF-I and increase its half-life both in the extracellular environment and the circulation [26]. Circulating IGF-I exists predominantly as a ternary complex, consisting of IGF-I, IGFBP-3 or IGFBP-5 and the glycoprotein acid-labile subunit (ALS). This complex retains a circulating reservoir of IGF-I and protects it from proteolytic degradation [21]. Moreover, in a ternary complex IGF-I is inactive, because in this form it is unable to cross capillary membranes or activate the receptor(s) [11, 19]. Hence, IGFBPs are expected to modulate the extent of IGF-dependent cellular effects, both in the circulation and in the extracellular matrix, via regulation of free IGF-I concentration and its local bioavailability in the tissue [20, 26]. Under stress conditions (e.g., trauma) the affinity of IGFBPs, and particularly of IGFBP-3, for IGFs is reduced by proteolytic clipping of IGFBP-3 and, thus, IGF-I can re-associate with lower molecular weight binding proteins in binary complexes, which enables it to cross capillary membranes and target the peripheral tissues [11]. On the other hand, IGFBPs compete with IGF-IR, as they normally have higher affinity to IGF-I than IGF-IR. Therefore, binding of IGFBPs to IGF-I prevents the ligand to interact with the receptor, thus suppressing IGF-I actions. The ratio between free IGF-I and IGFBP-IGF-I bound, as well as the tissue-specific distribution of particular IGFBPs control the IGF-I-inhibitory or stimulatory activities of IGFBPs [7, 27].
Further, it appears that the IGFBPs have also independent activity in modulating mitogenesis, cell survival and apoptosis [27, 28], while there is also a subgroup of binding proteins, known as IGFBP-related proteins, that exhibit low binding affinity to IGFs and their functions regarding the IGFs actions are yet unclear [11, 29].
11.2.3 IGF-I Receptors
IGF-I actions are mediated through its binding to specific cell surface receptors (R) already mentioned, i.e., the IGF-IR, IGF-IIR, IR, and the hybrid IGF-IR/IR receptors [19], (Fig. 11.2). More specifically, IGF-I binds IGF-IR with the highest affinity, IGF-IIR with low affinity and is also able to interact with IR, but with much lower affinity [1]. The IGF-I exhibits significant structural similarity to insulin and both can cross-activate IGF-IR and IR, while the IGF-IR signaling pathways share multiple intracellular mediators with the insulin signaling cascade [13]. Moreover, various domains in the IGF-IR have a high degree of similarity (40–84 % aminoacid sequence homology) to IR [3, 30], both of which are comprised of 2 α-subunits and 2 β-subunits linked by disulfide bonds. However, the affinity of insulin for the IGF-IR is about 100-fold less than that of IGF-I and, thus, high concentrations of insulin are needed to cause IGF-IR activation. Nevertheless, and despite the similarities in their intracellular signaling cascades, the IGF-IR and the IR elicit different responses, probably due to differences in their subcellular localization and in the structure within the kinase containing β-subunit, as well as due to ligand specificity and, thus, the different conformational changes conferred by the α-subunits (reviewed in Suleiman et al. [11]).
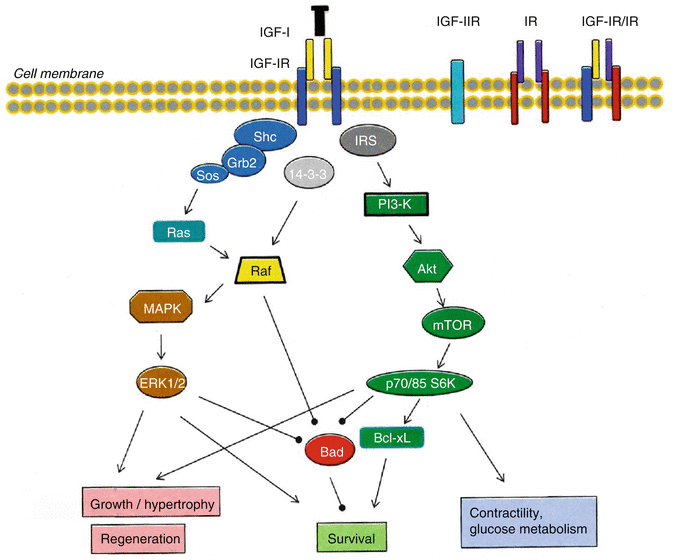
Fig. 11.2
Schematic representation of the insulin-like growth factor I (IGF-I) receptors, and IGF-I signaling pathways downstream of IGF-IR that result in specific biologic effects in myocardial cells
IGF-IR/IR hybrid receptors are present in many mammalian tissues including the heart. They are heterologous IGF-IR and IR α–β hemireceptors that maintain autophosphorylation activity upon ligand binding. The IGF-IR/IR hybrid receptor binds both insulin and IGF-I, however is thought to function predominantly as an IGF-I receptor, since its binding affinity for IGF-I is higher than that for insulin [21, 31]. The functional importance of IGF-IR/IR hybrid receptors remains poorly understood, however there is evidence that they may play an important role in both IGF-I and insulin-mediated cardiac growth (reviewed in DeBosch and Muslin [6]).
IGF-I acts primarily through the binding and activation of the IGF-IR. This is a transmembrane, ligand-activated receptor tyrosine kinase consisting of two extracellular α-subunits, which contain the cysteine-rich ligand binding site, and two transmembrane β-subunits which have a cluster of three tyrosine residues that undergo phosphorylation and thus activation upon IGF-I binding [1, 30, 32], (Fig. 11.2). Ligand binding to the IGF-IR causes a structural rearrangement in the transmembrane β-subunits of the receptor, resulting in trans-autophosphorylation of the tyrosine residues as one kinase domain phosphorylates the other, and thus destabilizing the auto-inhibitory conformation within the kinase domain of the receptor [33]. The IGF-IR activation leads to a trend in protein substrates in favor of the catalytic site, thus recruiting and phosphorylating specific cytoplasmic molecules and adaptor proteins, including Src homology/collagen (Shc) and insulin receptor substrate (IRS) proteins [2], (Fig. 11.2). IRS-1 and IRS-2 do not have an intrinsic kinase activity and act to recruit other enzymes, in order to elicit a variety of responses through specific intracellular signaling pathways. The IGF-IR, after its ligand-induced activation, is usually down-regulated by endocytic internalization [11].
11.3 IGF-I/IGF-IR Signaling and Biological Actions on the Myocardium
There is a growing interest in recent years regarding the role of IGF-I in cardiac physiology, largely within the context of cardiac repair and stem cell research, and numerous studies have implicated IGF signaling in the regulation of cardiac growth, homeostasis, viability and regeneration [3, 11, 34].
It is widely recognized that most of the observed IGF-I biological effects on cell proliferation, differentiation and survival depend on the activation of IGF-IR [7, 11]. Ligation of IGF-IR phosphorylates specific molecules and initiates intracellular signaling cascades involved in mitogenic, myogenic and cell-survival/anti-apoptotic activities [11, 34], (Fig. 11.2). IGF-I is produced locally in the heart and IGF-IR is also present in cardiac muscle [3]. Thus, based on the well known IGF-IR cellular signaling, IGF-I actions on cardiomyocytes are expected to include an increase in cell size, prevention of apoptosis, regulation of glucose metabolism and intracellular Ca2+, and direct effects on cardiac muscle contractile function [11, 35]. A variety of biological effects is regulated by the IGF-IR-induced stimulation of several phosphorylation cascades. Site-directed mutagenesis has been used to map specific signaling pathways, functions, and phenotypes associated with IGF-IR signaling, and receptor domain-dependent functions were identified for protection from apoptosis, anchorage-independent growth (i.e., cells grow in suspension or soft media where they float instead of growing on a solid substratum), and DNA synthesis (reviewed in Kurmasheva and Houghton [9]). Specific IGF-I/IGF-IR-dependent signaling and biological actions in myocardium are discussed in the next sections, (Figs. 11.2 and 11.3).
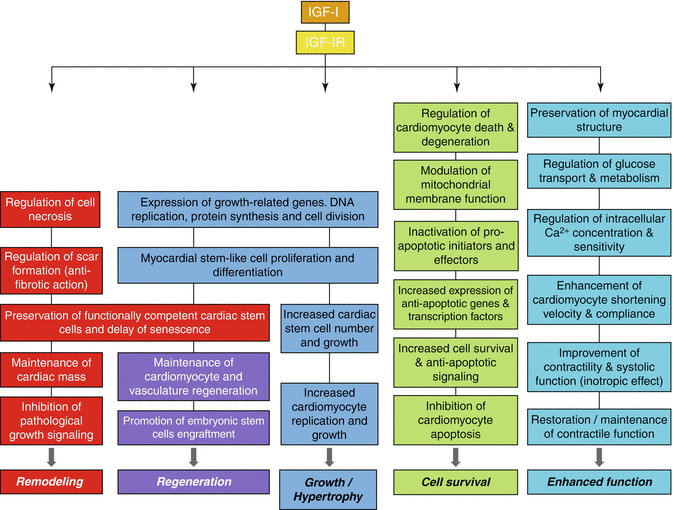
Fig. 11.3
Cellular processes and biologic effects in myocardium induced by the insulin-like growth factor I (IGF-I) binding to type 1 IGF receptor (IGF-IR)
11.3.1 IGF-I Signaling in Cardiac Development and Growth/Hypertrophy
Heart growth is often referred to as cardiac hypertrophy (H) and can be defined as physiological or pathological; physiological H includes heart growth during postnatal development (sometimes also called eutrophy) and in response to exercise training (as occurs in elite aerobic exercise athletes) and is associated with preserved or enhanced cardiac function. Pathological cardiac H occurs in disease settings such as myocardial infarction, hypertension and valve disease, and is typically associated with histopathology and depressed cardiac function. The activation of distinct signaling transducers causes the different phenotypes of cardiac hypertrophy [36, 37].
That the IGF system plays an important role in the development and growth of the heart [11], GH/IGF-I axis may influence myocardial growth [38], and GH/IGF-I deficiency may lead to cardiac atrophy and impaired cardiac function [39]. More specifically, IGF-I appears to be crucial for the embryonic growth with age of the myocardium and it has an important role in its response [3]. Physiological cardiac hypertrophy in athletes is also associated with increased cardiac IGF-I production. This growth factor has been shown to have specific cardiac effects, stimulating DNA and protein synthesis, cardiomyocyte proliferation, differentiation and H [37, 40], and has been associated with the induction of expression of contractile proteins in neonatal rat cardiomyocytes [40], for review see [3], (Fig. 11.3).
Although substantial data suggest that IGF-I is a potent cardiomyocyte growth factor, nevertheless the research data regarding the IGF-I-induced myocardial hypertrophy appear controversial [11]. Specifically, IGF-I overexpression in mice increased cardiac stem cell number and growth [41], while myocardium-specific overexpression of IGF-I resulted in increased number of cardiomyocytes and total heart weight [42]. Moreover, it has been reported that overexpression of IGF-I in the hearts of transgenic mice induced a hypertrophic phenotype [43], in other studies transgenic mice overexpressing IGF-IR in the heart exhibited increased cardiomyocyte size, enhanced contractile function and no evidence of histopathology [44, 45]. However, other studies failed to find cellular hypertrophy in cardiomyocytes in vitro, or in individual muscle fibers in the hearts of IGF-I overexpressing mice, or of mice overexpressing IGF-I specifically in the heart [42, 46], reviewed in Suleiman et al. [11].
The cardiac growth/hypertrophy-promoting actions of IGF-I are mediated through various IGF-IR signaling components, involving extracellular signal-regulated kinases (ERKs), IRS-1 and phosphatidylinositol 3-kinase (PI3-K) [2, 34, 37], (Fig. 11.2). Specifically, mitogenic IGF-I signaling cascades are activated through the phosphorylation of Shc, which connects IGF-IR tyrosine kinases to mitogen-activated protein kinases (MAPK) through the Ras-Raf-MEK-ERK signaling pathway. Shc binds to phosphorylated tyrosines on activated IGF-IR and its subsequent phosphorylation generates a binding site for the growth receptor binding protein 2 (Grb2) and subsequently a Shc/Grb2/Son of Sevenless (Sos) complex. Sos activates the small G protein Ras, which is located at the plasma membrane and in its turn associates with and activates protein serine kinase Raf [3, 13, 34], (Fig. 11.2). The activation of Ras/Raf signals to MKKs (specifically MEK1/2) and these kinases phosphorylate on specific sites (Thr-Glu-Tyr) and activate ERKs (ERK1/2), which then can activate by phosphorylation other protein kinases and several transcription factors [34].
Although the Ras-Raf-MEK-ERK signaling pathway appears to regulate certain intracellular hypertrophic responses, however the importance of this pathway in the regulation of cardiac H has been disputed [34, 47]. Specifically, both in vivo and in vitro studies have shown that acute exposure of rat adult ventricular cardiomyocytes and the cardiomyoblast cell line H9C2 to IGF-I resulted in the activation of the IGF-IR, followed by the sequential activation of the Ras-Raf-MEK-ERK cascade, which is required for the anabolic effects of IGF-I [11]. In addition, it was demonstrated that ERK signaling is necessary for phenylephrine-induced cardiomyocyte H in culture [48], and that ERKs are required for sarcomeric organization induced by hypertrophic agonists, suggesting a more specialized role in cardiomyocyte H [49]. Moreover, agonist stimulation or cell stretching resulted in the activation of ERK1/2 in cultured cardiomyocytes [50], implicating ERK1/2 signaling proteins as regulators of a hypertrophic response. Nevertheless, there are also studies suggesting a minimal role for ERKs in cardiac hypertrophy [51]. Overall, although the necessity of ERK signaling as a hypertrophic mediator is disputed, it appears that ERKs are implicated as downstream effectors of the hypertrophic response in the myocardium [34].
Other signaling molecules that appear to regulate the physiological hypertrophy phenotype in the myocardium are the components of the IGF-I/PI3-K/Akt pathway [35–37]. In particular, the regulatory subunit of PI3-K contains a src homology 2 (SH2) domain which interacts with IRS-1, resulting in PI3-K activation [36]. PI3-K then leads to the activation of Akt (protein kinase B) as well as p70S6K and p85S6K, which affect diverse intracellular processes such as translational regulation, cardiac cell growth/hypertrophy and survival (discussed in the next section), [34, 35, 52]. IGF-I receptor tyrosine kinases activate the PI3-K(p110-alpha) pathway leading to physiological H, whereas pathological H involves the activation of PI3-K(p110-gamma) pathway induced by G-protein coupled receptors (GPCRs) [12, 53]. Cardiac-specific overexpression of a constitutively active PI3-K catalytic subunit (p110) in mice resulted in cardiac H, which was associated with a comparable increase in myocyte size, while the changes in heart size were correlated with PI3-K activity. Moreover, transgenic overexpression of a dominant negative PI3-K resulted in smaller hearts and myocytes [54].
One of the most comprehensively studied signaling molecules in the regulation of cardiac growth and function is Akt kinase [12, 55]. Akt is downstream of the IGF-I/PI3-K/Akt physiological H pathway and it regulates a range of downstream targets involved in the modulation of local responses. It also regulates processes such as cell growth, differentiation and survival, as well as cell metabolism and contractile function [12, 55], (Fig. 11.2). Studies utilizing various Akt mutant mouse models suggest that the subcellular distribution of Akt activity is a determinant for the resulting cardiac phenotype. Moreover, short-term activation of Akt induces physiological H, whereas its prolonged activation results in pathological H, indicating that, depending on the timing and extent of stimulation, the IGF-I/Akt pathway may sustain either physiological or pathological hypertrophy [12, 56]. Indeed, Akt is critical both at baseline and for hypertrophic adaptation and the activation of the IGF-I/PI3-K/Akt pathway plays an important role in the mechanism of physiological cardiac H, by promoting a coordinated angiogenic program. Overall, physiological H is associated with the activation of the IGF-I/PI3-K/Akt pathway, which is critical in modulating cardiac growth and vasculogenesis (reviewed in Catalucci et al. [12], see also next Sect. 11.3.3).
In addition, there is evidence that the IGF-I/PI3-K/Akt signaling pathway downstream effectors, p70S6K and p85S6K, may also play a role in regulating physiologic cardiac H. Ventricular pressure overloading resulted in the activation of both p70S6K and p85S6K [52], while selective inhibition of p70S6K blocked the augmentation of agonist-induced protein synthesis and the subsequent hypertrophic response of cultured cardiomyocytes [34, 57].
11.3.2 IGF-I Signaling in Cardiac Cell Survival/Inhibition of Apoptosis
Programmed cell death apoptosis (APO) is a key contributor to myocyte loss that accompanies many forms of myocardial disease, such as acute myocyte death after ischemic injury. APO in cardiomyocytes contributes to the development of heart failure [58], while it may also occur even during normal aging. It has been demonstrated that the constitutively expressed biochemical apoptotic machine in cultured cardiomyocytes can be suppressed by the anti-apoptotic signals of IGF-I in the serum [59].
IGF-I has been well characterised as an anti-apoptotic factor and in many respects it appears to be a more potent survival than mitogenic factor. The identity of all the signaling proteins and interactions that mediate the IGF-I survival effect remains to be fully elucidated, however many components have been identified [11]. Thus, IGF-I is a potent cardiomyocyte survival factor and may affect the cardiac muscle mass by preventing APO and cardiomyocyte loss [11, 60]. Indeed, IGF-I has protective effects on the heart, as cardiac-specific IGF-I overexpression is anti-apoptotic [41] and results in reduced reperfusion injury and less cardiomyocyte death and fibrosis in chronic coronary artery disease [60], (Fig. 11.3). On the other hand, IGF-I-deficient mice have been shown to exhibit increased APO after myocardial infarction [61]. Moreover, IGF-I can decrease myocyte APO and prolong cell survival in cultured cardiomyocytes, and the anti-apoptotic effects of IGF-I on cardiac muscle cells can occur at physiological concentrations [59, 62]. Short-term administration of IGF-I in animals has been reported to counteract APO and improve cardiac contractility, however clinical trials in which IGF-I was administered chronically have shown conflicting results [3, 12, 44].
Several signaling pathways have been suggested to mediate the anti-apoptotic effects of IGF-I [63]. Specifically, three different pathways, originating from different domains of the IGF-IR, are involved in cell survival: the IRS-1/PI3-K/Akt/p70S6K pathway, acting through the tyrosine kinase domain; the Shc/MAPK pathway, acting preponderantly through the Y950 domain; and a third pathway which is dependent on the serine cluster motif (S1280–1283) of the IGF-IR and acts in cooperation with 14-3-3 proteins. The latter pathway results in the translocation of Raf-1 to mitochondria where it can phosphorylate, and so inactivate, the pro-apoptotic protein Bad, a member of the Bcl-2 family of proteins, (Fig. 11.2). Indeed, IGF-I acts as a survival factor via the stimulation of the Bcl-2 proteins [8], as IGF-I-mediated inhibition of APO has been shown to be associated with increased expression of an anti-apoptotic member (Bcl-xL) of the Bcl-2 family of proteins [64]. Mitochondria appear to have an important role in the apoptotic death and IGF-I has been shown to modulate mitochondrial membrane function through activation of PI3-K/Akt pathway [65]. The operation of any two of the above three pathways is sufficient to protect cells from APO, while inactivation of Bad appears to be a target for each pathway [9, 13], (Fig. 11.2).
The most frequently implicated pathways in IGF-induced survival signaling are MAPK and PI3-K pathways. IGF-IR signaling via IRS-1, Akt or MAPK regulates positively the expression of anti-apoptotic genes and transcription factors that themselves regulate anti-apoptotic programs, and negatively regulates the expression of pro-apoptotic genes. The requirement of PI3-K or ERK1/2 in mediating IGF-survival appears to be depended on the model system and the stress stimulus used. Inhibition of PI3-K or MAPK ablates the survival effect of IGF-I and under serum starvation conditions activation of both PI3-K and ERK1/2 is required, indicating that both are vital components of the anti-apoptotic effects of IGF-I [9, 11, 63]. In the absence of IRS proteins, sustained activation of ERK1/2 appears largely dependent on the Y950 domain, although the relationship between IGF-I-mediated survival and activation of ERKs is not absolute [9].
The primary cell survival pathway activated by IGF-I is the PI3-K/Akt signaling pathway (Fig. 11.2), which is activated in the presence of IRS-1 [9]. Tyrosine kinase-phosphorylated IRS-1 and IRS-2 interacts with specific cytoplasmic proteins, leading to the transduction of downstream cell survival and anti-apoptotic signals [33]. IRS proteins may play differential roles in the anti-apoptotic effects of IGF-I with IRS-1 being predominant. IRS-1 appears to be important in mediating IGF-I protection from APO under serum-starved conditions, as PI3-K activity and Akt phosphorylation were found to be reduced in IRS-1 deficient cells [9]. Phosphorylation of PI3-K activates the Akt pathway which inhibits activation of pro-apoptotic initiators, such as Bad, and effectors, e.g., caspases [8, 11, 36], while inhibition of PI3-K signaling would prevent the completion of the cell cycle, leading potentially to cell APO or differentiation. Moreover, it has been shown that the PI3-kinase/Akt pathway protects cells from reperfusion injury and promotes cardiomyocyte viability both in vitro and in vivo [66], and Akt appears to be a critical component of the survival pathway, regulating the activities of apoptotic factors [9, 59, 67]. Overall, IGF-I/PI3-K pathway provides cardiac protection and the mechanisms responsible for this protection include its anti-apoptotic and anti-fibrotic properties, inhibition of signaling cascades that lead to pathological growth, and maintenance of contractile function in a disease setting and the regeneration (reviewed in McMullen [36]), (Figs. 11.2 and 11.3).
11.3.3 IGF-I Signaling in Cardiac Regeneration/Remodeling
There is increasing evidence for new myocyte formation in the adult heart and a growing interest in the extent and mechanisms of cardiomyocyte regeneration (REG) [68–70]. A large number of cardiomyocytes in mitosis and a high frequency of cardiomyocyte replication in normal hearts have been observed both in response to increased work load and in acute or chronic cardiac failure of ischemic and non-ischemic origin, both in animals and humans. These findings indicate that cardiomyocyte REG is an important contributor to the maintenance of cardiac mass in physiological and pathological conditions [68–70]. Myocyte death and REG are part of the normal homeostasis of the heart and the generation of new cardiomyocytes predominates over cell death and contributes significantly to the normal growth of the heart into adulthood [53]. Thus, although the adult heart is mainly composed of terminally differentiated cells, it should not be considered as a post-mitotic organ, since it exhibits a significant capacity for myocyte REG that is markedly enhanced in acute and chronic heart failure. In human end-stage ischemic hearts and in idiopathic dilated cardiomyopathy, a high increase in the number of mitotic cells has been reported compared to control hearts [68]. Nevertheless, human and animal studies have provided evidence that in the failing adult heart the magnitude of cardiomyocyte replication and REG is far below the extent of the myocyte death [62].
The cardiac regenerative capacity is supported by a subpopulation of cycling, myocardial stem-like cells which possess the potential of regenerating the muscle cells and the coronary vasculature of the myocardium [70, 71]. Whether the origin of the cardiac stem cells permanently resides in the myocardium or they are taken up from the circulation/bone marrow is not clear [71].
IGF-I appears not only to protect myocardial cells from death and degeneration, but also to be an important stimulant for the REG of the adult heart, by promoting the expression of growth-related genes, DNA replication and cell division [10], (Fig. 11.3). Specifically, IGF-I has been shown to promote myocyte renewal and myocardial REG in experimental protocols of ischaemic cardiac injury [72, 73]. In addition, intracoronary injections of IGF-I and hepatocyte growth factor (HGF) were capable of activating cardiac stem cells and subsequently regenerating new myocytes and vasculature lost after myocardial infarction in pigs. This myocardial REG was accompanied by improved cardiomyocyte survival and ventricular function [73], while an increase in cell number in IGF-I transgenic animals has been also observed, suggesting that IGF-I may play a role in the REG of the myocardium [36]. Moreover, cardiac stem cell division in IGF-I transgenic mice has been shown to be induced via the IGF-IR, and it was accompanied by preservation of a reservoir of functionally competent cardiac stem cells and delayed senescence [41], (Fig. 11.3).
The REG capacity of IGF-I has been particularly explored in transgenic mice overexpressing a class 1 IGF-IEa isoform (see previous Sect. 11.2.1), implicating it as a powerful mediator of the REG response [72]. In particular, this isoform has been reported to have regenerative properties and to promote cell survival and renewal and its expression in cardiac myocytes induced a hypertrophic response [74]. The IGF-IEa transgene was reported to induce repair of the injured tissue, by promoting regenerative properties in damaged heart tissue with minimal scar formation and restoring of cardiac function through increased anti-apoptotic signaling [72, 74].
In conditions such as myocardial infarction, ischemia/reperfusion injury and heart failure, there is often myocyte loss, although a later increase in myocardial mass may occur through compensatory myocyte hypertrophy/remodeling and/or increased collagen deposition (fibrosis) [58]. Thus, after the segmental loss of myocardium due to ischemic heart disease, myocyte REG and H processes are activated contributing together to the development of new cardiac muscle mass. However, both processes are restricted to the remaining viable myocardium and the border zone of the infarct, while a scar formation occurs in the healed infarcted area [70]. The urokinase-type plasminogen activator (uPA)/transforming growth factor beta 1 (TGF-β1) bioregulation system appears to have an important role as a main regulator of the myocardiac collagen accumulation and fibrosis, and the extracellular matrix remodeling [12, 75, 76].
Interestingly, intracoronary injections of IGF-I/HGF have been proven effective to reduce the pathological cardiac remodeling (RΕΜ) and fibrosis after myocardial infarction [53]. In addition, the influence of IGF-I on myocardial remodeling involves also an adaptation of the coronary vasculature [11]. Thus, increased expression of IGF-I has been observed in rat aortas following balloon injury [77]. Moreover, in a variety of disorders such as hypertrophic cardiomyopathy, “hypertensive” heart disease and “ischaemic” cardiomyopathy, there is an initial up-regulation of IGF-I and IGF-IR, which has been suggested to be involved in a cardiac remodeling response [3, 11], (Fig. 11.3). Besides, higher serum IGF-I levels immediately after the onset of acute myocardial infarction have been associated with less myocardial remodeling and improved ventricular function [78], (see discussion in the next section).
11.3.4 IGF-I Signaling and Cardiac Function
IGF-I affects cardiac functional changes by improving cardiac contractility, stroke volume, cardiac output and ejection fraction [3], although enhanced cardiac contractility and elevated blood pressure have been reported in IGF-I-deficient mice, indicating that IGF-I deficiency can selectively modulate blood pressure and left ventricular function, yet normal IGF-I levels are not required for adaptive myocardial hypertrophy in response to sustained hemodynamic load [79]. Chronic heart failure (HF) leads to the appearance of cardiac cachexia in the late stages of the disease [80], which is accompanied by increased rate of APO and reduced local expression of IGF-I. There is strong evidence that a low IGF-I level increases the risk of HF. In patients with HF, IGF-I levels are low and have been correlated with left ventricular mass and ventricular systolic dysfunction [39]; GH/IGF-I deficiency may lead to cardiac atrophy and impaired cardiac function [3, 39]. Moreover, it has been shown that serum IGF-I concentrations at the time of cardiac infarction in humans can be used to predict later development of HF [62, 78]. In addition, in elderly people without a previous myocardial infarction, serum IGF-I levels were found to be inversely related to the risk for congestive HF [81], while the relative risk of coronary artery disease mortality has been associated with decreased levels of circulating IGF-I [82]. Besides, IGF-I can decrease systemic vascular resistance resulting potentially in indirect effects on the heart [83], while intravascular infusion of IGF-I resulted in significant increase in regional forearm blood flow when administered to humans [84], implying that IGF-I may be involved in the regulation of vasodilatation [11].
It has been suggested that IGF-I administration could benefit the cachectic heart, since this growth factor increases cardiac stem cell numbers and growth and leads to an increase in myocyte turnover and function [11, 41]. Indeed, intracoronary administration of IGF-I has been shown to significantly improve myocardial function during the early phase of myocardial infarction in pigs, and this functional improvement was associated with the preservation of myocardial structure [85]. Nevertheless, chronic infusion of IGF-I has been shown to result in mild cardiac hypertrophy in rats [86], and sustained overproduction of IGF-I may lead to hypertrophic cardiomyopathy [3]. In addition, local IGF-I overexpression in skeletal and cardiac muscle has been shown to initially induce first physiological and later pathological cardiac H in transgenic mice [43]. Collectively, it has been suggested that both extreme IGF-I overproduction and deficiency may lead to deterioration of cardiac function [62], (see also discussion below).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


