The Medical Management of Chronic Heart Failure in Patients with Systolic Dysfunction
Barry H. Greenberg
Since publication of the first edition of this book, substantial changes in the medical management of heart failure have profoundly altered the clinical course of patients with this condition. Although our ability to define exactly the mechanism by which a treatment affects the natural history of heart failure is more limited than we sometimes imagine, new approaches to therapy have generally been based on improved understanding of the underlying pathophysiology of heart failure. Well-designed clinical trials have been the testing ground for new therapies and, as such, their results form the basis for most of the recommendations presented in this chapter. Ironically, as we learn more about heart failure and the patients who suffer from this condition, we have come to realize that clinical trial results may have limited applicability. This is due to the fact that the heart failure population is extremely heterogeneous and that many important groups of patients, most notably females, the very elderly and, in particular, patients with significant comor-bidities, are underrepresented in clinical trials. Nonetheless, well-designed clinical trials provide proof of concept and they remain the ultimate rationale for changes in treating heart failure. Based on the results of these trials, there is much reason for optimism for patients with this condition since there is compelling evidence to indicate they have enjoyed substantial gains in both the quantity and quality of their lives due to these new therapies.
The goal of this chapter is to provide an integrated approach to the medical management of patients with heart failure due to systolic dysfunction. The treatment of patients who develop heart failure with preserved ejection fraction (EF) is reviewed in a separate chapter devoted to this entity alone. Since heart failure patients may or may not have signs and symptoms of fluid overload, the term chronic heart failure rather than congestive heart failure will be used throughout. Other chapters in this text include extensive reviews of the pathophysiology, prognosis, and drugs used to treat chronic heart failure, and this chapter will focus on selected aspects of these topics only as they relate to a rational and integrated approach to therapy. Medical management requires an understanding of the pathophysiological framework, and the chapter begins with a brief overview of mechanisms involved in the development and progression of heart failure. It is followed by a review of the drugs (diuretics, angiotensin-converting enzyme inhibitors, β-blockers, digoxin) that are the cornerstones of therapy as well as other agents for which information demonstrating efficacy has become available over the past few years (aldosterone antagonists, angiotensin receptor blockers, and the combination of hydralazine and isosorbide dinitrate).
Another major addition to this chapter is the discussion of the role of device therapy in the treatment of patients with chronic heart failure. Advances in this area have been substantial since the last edition of the text and it is imperative that these new approaches to treating heart failure be considered in the context of the pharmacologic therapies to which they are added in order to maximize their benefits. The results of recent clinical trials will be emphasized since information derived from these studies drives the new directions in management of heart failure patients that are outlined in this chapter. Practical aspects of therapy will also be covered and recommendations for the treatment of patients with varying stages of heart failure will be presented.
Pathophysiology
In order to effectively approach the many issues related to the treatment of the chronic heart failure patient, it is first necessary to define the condition. This task has become progressively more difficult as increased sophistication of the various disciplines and exigencies of funding have widened the chasm between basic researchers and practicing clinicians. Nonetheless, the heart failure phenotype that develops in genetically modified mice and other experimental animal models is not so different from what the clinician encounters when faced with the patient who presents with a large, poorly contracting ventricle, rales, and pedal edema. A working definition that might satisfy both constituencies is that chronic heart failure is a syndrome in which the inability of the heart to provide adequate amounts of oxygenated blood to vital tissue at normal levels of ventricular filling pressure provokes a widespread and progressive response that affects the structure and function of the heart and other organs throughout the body. One key aspect of this definition is that chronic heart failure is a syndrome that develops as the end result of a variety of different disease processes. Some of these diseases may not even primarily affect the heart. Furthermore, this definition calls attention to the fact that chronic heart failure is a dynamic and progressive process that eventually alters the physiology of the entire organism.
As outlined in Table 35-1, heart failure can be attributed to both cardiac and noncardiac causes. Among the cardiac etiologies, myocardial dysfunction is the most common. Myocardial abnormalities that primarily affect systolic function and those that predominantly alter diastolic function can both give rise to the syndrome of heart failure. Most commonly, however, systolic and diastolic dysfunction exist in the same patient. Valvular heart disease, congenital or acquired structural abnormalities, and pericardial disease are all primarily nonmyocardial causes of heart failure. In each of the latter, however, secondary changes in myocardial structure and function are usually involved and may continue to cause chronic heart failure, even after the primary structural abnormality is corrected.
The pathogenesis and management of heart failure on the basis of diastolic myocardial dysfunction, as well as extracardiac and nonmyocardial causes, are covered in Chapters 19, 20, and 22. Recognition of the fact that heart failure is a syndrome with multiple etiologies, however, brings up one of the most important principles of management. That is, in order to decide on a rational course of therapy, it is essential to define as best as possible the cause of the syndrome and to determine whether or not there is a treatable (and even potentially reversible) etiology. Clearly, the approach to the patient with chronic heart failure due to long-standing severe anemia or on the basis of mitral stenosis will be dramatically different than in a patient who develops progressive cardiac dysfunction due to remodeling after myocardial infarction (MI) or as consequence of doxorubicin myocardial toxicity.
A characteristic of chronic heart failure due to myocardial systolic dysfunction is that at some point, either at rest or during exercise, there is a reduction in the capacity of the heart to pump blood. Regardless of the etiology, the resulting reduction in blood pressure and/or cardiac output stimulates a pathophysiological response involving the heart and multiple other organs throughout the body. The primary goal of the compensatory changes, which are summarized in Table 35-2, is to augment the delivery of oxygenated blood to vital organs such as the heart and brain. It is extremely important to recognize that, in addition to providing (mostly short-term) support for the failing heart, these compensatory changes are also associated with effects whose deleterious nature become apparent over time. For instance, retention of salt and water and venoconstriction serve the important function of increasing blood return to the heart. This helps maximize stretch of cardiac sarcomeres. The resulting increase in shortening of these contractile units will augment stroke volume. The increases in left ventricular (LV) filling pressure and volume that occur, however, contribute to the development of congestive
symptoms. They also increase LV wall stress. In addition to increasing myocardial oxygen demands, the increase in wall stress activates mechanoreceptors and stimulates local neurohormonal release, both of which promote cardiac remodeling (1,2,3,4,5,6). In this manner, fluid retention ultimately leads to both symptoms of heart failure and progression of the underlying abnormalities in cardiac structure and function.
symptoms. They also increase LV wall stress. In addition to increasing myocardial oxygen demands, the increase in wall stress activates mechanoreceptors and stimulates local neurohormonal release, both of which promote cardiac remodeling (1,2,3,4,5,6). In this manner, fluid retention ultimately leads to both symptoms of heart failure and progression of the underlying abnormalities in cardiac structure and function.
Table 35-1 Conditions Resulting in the Syndrome of Chronic Heart Failure | ||
|---|---|---|
|
Table 35-2 Compensatory Changes In Chronic Heart Failure | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
An aspect of heart failure that is now well recognized is the fact that once myocardial damage occurs, changes in cardiac structure and function are not static. Even when the process is initiated by a discrete event such as an MI, there is convincing evidence that deterioration in myocardial systolic function is progressive (7,8,9,10). This is due to the process of cardiac remodeling which occurs in response to injury and/or to alteration in global and regional myocardial loading conditions. In these settings neurohormonal systems are activated throughout the body as well as in the heart itself (11,12,13,14,15,16). There is convincing evidence that many of these neurohormonal systems promote maladaptive cardiac remodeling. Moreover, we now recognize that by blocking the renin-angiotensin system (RAS), aldosterone, and the sympathetic nervous system we can prevent (or reverse) LV remodeling and systolic dysfunction, relieve symptoms, improve quality of life, and significantly reduce heart failure morbidity and mortality.
Drugs Used in the Treatment of Chronic Heart Failure
Until recently, diuretics, digoxin, and the angiotensin-converting enzyme (ACE) inhibitors were considered to be the three cornerstones of therapy for heart failure. Information collected over the past decade has allowed β-blockers to enter into this therapeutic pantheon. In addition, there is now information that (at least) selected groups of patients may also greatly benefit by the addition of an aldosterone antagonist, an angiotensin receptor blocker, or the combination of hydralazine and isosorbide dinitrate to their therapeutic regimen.
Diuretics
Because patients with heart failure almost always present with signs and symptoms of pulmonary or systemic congestion, diuretic agents play an integral role in the therapeutic
approach to this syndrome. Although there is only limited evidence from clinical trials to document their efficacy (17,18) and no clinical trials demonstrating that they improve survival, the obvious impact of diuretic agents on the signs and symptoms of heart failure has confirmed their therapeutic value in a manner analogous to the use of penicillin in treating pneumococcal pneumonia.
approach to this syndrome. Although there is only limited evidence from clinical trials to document their efficacy (17,18) and no clinical trials demonstrating that they improve survival, the obvious impact of diuretic agents on the signs and symptoms of heart failure has confirmed their therapeutic value in a manner analogous to the use of penicillin in treating pneumococcal pneumonia.
Although diuretics are an essential component of the therapeutic approach to chronic heart failure, it is important to recognize that they are effective primarily as a means of removing excess salt and water. As indicated in Table 35-2, patients with chronic heart failure retain fluid in order to increase LV stroke volume. This response, however, is excessive in most instances. As shown in Figure 35-1, the high levels of LV end-diastolic volume that are usually seen in chronic heart failure are no longer effective in enhancing cardiac output because the patient is positioned on the flat portion of the LV function curve. The accompanying increase in LV filling pressure, however, leads to the development of the signs and symptoms of congestion. The predominant impact of diuretic therapy is to reduce filling pressure and relieve congestion. In this case, the patient moves from point A to point B in the figure. In some instances, diuretic therapy also may increase cardiac output (19,20). This is depicted in Figure 35-1 by movement to point D on a new curve that is positioned upward and to the left. There are several mechanisms through which this can occur. Perhaps the most important is that patients with secondary mitral regurgitation (on the basis of dysfunction of the subvalvular apparatus) may experience a considerable improvement in valvular competence as LV volumes are reduced. Cardiac output also can be increased on the basis of a reduction in wall stress (i.e., afterload) due to a decrease in chamber radius or when relief of pulmonary congestion leads to diminished neurohormonal activation and a consequent reduction in peripheral vasoconstriction (19). Finally, reductions in wall stress help reduce myocardial oxygen demands. In situations where myocardial ischemia is present, the favorable effect that this would have on the balance between myocardial oxygen supply and demand would tend to improve LV contractile performance and increase cardiac output.
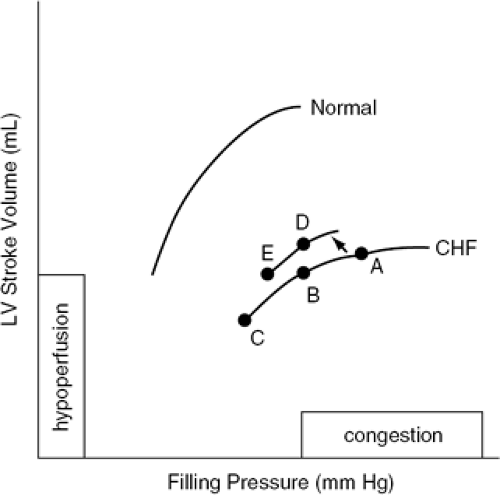 Figure 35-1 Representative LV function curves from a normal subject and a patient with heart failure. |
Excessive use of diuretics, however, can lead to a reduction in cardiac output. This is depicted in Figure 35-1 by movement to point C or point E and is related to the fact that diuretics can move the patient from the flat portion of the LV function curve to the ascending portion. In this position, stroke volume is again responsive to the preload stretch of the sarcomeres, and as filling volume is further decreased, cardiac output is reduced. This situation tends to occur when the patient is being aggressively diuresed. Overdiuresis also can be seen as an insidious late occurrence in patients whose LV systolic dysfunction improves over time either spontaneously or in response to therapy. It also tends to occur not infrequently in warm and humid environments when the amount of insensible fluid loss increases. When hypoperfusion due to excessive diuresis occurs, it can be recognized clinically by the complaints of increased weakness and fatigue, signs and symptoms of postural hypotension, or an increase in blood urea nitrogen (BUN) that is out of proportion to increases in serum creatinine (i.e., the BUN/creatinine ratio is increased). When this occurs, reduction in the dose of diuretic is required.
As a result of their potency, relatively low side effect profile, and ability to affect water and solute loss even at low levels of glomerular filtration, loop diuretics such as furosemide, bumetanide, and torsemide are most commonly used for the treatment of heart failure (21,22). Diuretics that act on other portions of the nephron may be used in association with one of the loop diuretics to accomplish specific purposes. For instance, a thiazide diuretic such as metolazone, which acts on the distal collecting tubule, can be used with a loop diuretic to help enhance diuresis in otherwise resistant patients (23,24). In other instances, a potassium-sparing diuretic can be used with a loop diuretic when potassium supplementation proves inadequate to maintain serum levels within the normal range. As will be discussed, the use of an aldosterone antagonist in this setting is particularly appealing since it is associated with a significant reduction in mortality, at least in patients with post-MI left ventricular dysfunction or with advanced heart failure symptoms.
The goal of diuretic therapy is to relieve the signs and symptoms of salt and water retention. In most cases diuretic therapy is initiated along with recommendations to reduce daily sodium intake to the range of 2 g. Even greater reductions to 1.5 g of sodium may be necessary in some clinically tenuous patients. Once patients achieve the euvolemic state, diuretics still need to be continued. The dose, however, may need to be lowered to avoid overdiuresis. Diuretic unresponsiveness can occur owing to a variety of causes, the most common being a deterioration in heart or kidney function; bowel edema, which alters drug absorption; or the use of drugs such as nonsteroidal antiinflammatory agents, which inhibit the natriuretic effects of loop diuretics (25,26). Diuretic resistance should be treated by first defining the cause. The use of combined diuretic therapy (as described above) or the temporary administration by the intravenous route may be needed to
overcome diuretic resistance. In some instances when this condition is due to a reduction in cardiac output and/or renal perfusion pressure, inotropic support with dobuiamine or milrinone or the use of ultrafiltration may be necessary to effectively diurese an otherwise refractory patient.
overcome diuretic resistance. In some instances when this condition is due to a reduction in cardiac output and/or renal perfusion pressure, inotropic support with dobuiamine or milrinone or the use of ultrafiltration may be necessary to effectively diurese an otherwise refractory patient.
Although diuretics are of unquestionable value in patients who have signs and symptoms of volume overload, their impact on the overall clinical course has been called into question. A post hoc analysis of the Studies of Left Ventricular Dysfunction (SOLVD) database found that compared with patients not taking any diuretic, the risk of hospitalization or death due to worsening HF in patients taking non-potassium-sparing diuretics (PSDs) alone was significantly increased (risk ratio [RR] = 1.31, 95% CI 1.09-1.57; p = 0.0004); this was not observed in patients taking PSDs with or without a non-PSD (RR = 0.99, 95% CI 0.76-1.30; p = 0.95) (27). Whether this effect was related to a direct action of the diuretic or to other factors that were not adequately controlled for in the course of the analysis cannot be known for certain.
However, these results raise the possibility that loop diuretics might have opposite effects in heart failure patients by relieving symptoms while at the same time adversely affecting long-term prognosis. The latter possibility could be due to diuretic induced electrolyte abnormalities, enhanced neurohormonal stimulation, or an increase in cardiac fibrosis. While the validity of this hypothesis remains uncertain, it does serve as a reminder that diuretic agents should not be given except to help remove accumulated salt and water or to lower elevated blood pressure in the heart failure patient. It is prudent to periodically reassess the need for diuretics and to reduce their dose or even discontinue in stable euvolemic patients. When the latter is being considered, it is important to establish the patient’s reliability in recognizing signs and symptoms of volume overload and their ability to restart the diuretic if necessary.
Diuretic agents should never be used as monotherapy in patients with heart failure. As mentioned previously in this chapter, the predominant effect of these drugs is to relieve congestive symptoms. When and if improvements in cardiac function occur, they tend to be modest. Diuretic agents do not alter the natural progression of cardiac dysfunction due to remodeling. Moreover, diuretics activate neurohormonal systems (particularly, the RAS) that have been implicated as causative factors in the progression of disease (28). Thus, diuretics would not be expected to halt or reverse cardiac remodeling and they always must be given in association with neurohormonal blocking agents, as outlined later.
Diuretic agents are associated with a variety of important side effects. The metabolic and electrolyte abnormalities that occur, particularly those involving renal function or serum potassium levels, are a major concern and require careful clinical attention as well as the judicious use of blood chemistry analyses in the follow-up management of patients. Hypokalemia is an important concern because of its association with ventricular arrhythmias (29,30). Thus, use of supplemental potassium is an important adjunct to diuretic therapy in most patients. The use of ACE inhibitors, angiotensin receptor blockers, or aldosterone antagonists tends to decrease the likelihood of hypokalemia, whereas the use of metolazone in association with a loop diuretic tends to increase the likelihood of hypokalemia. The dose of potassium supplementation often needs to be modified when one of these agents is used. Addition of a potassium-sparing diuretic or correction of hypomagnesemia may be needed when refractory hypokalemia occurs.
Digitalis Glycosides
It remains one of the great oddities of clinical medicine that it has taken over two centuries to determine whether the digitalis glycosides are effective therapy for chronic heart failure. The debate has had temporal aspects, with enthusiasm for the drug running high during certain eras but not in others, and there have been geocultural factors leading to widespread acceptance in countries such as Germany and profound skepticism in England. It would seem to be a relatively straightforward process to determine if a particular agent or class of agents does or does not improve the well-being of patients with chronic heart failure. After all, these very issues were decided in relatively short order for the ACE inhibitors (which are helpful) and for oral milrinone (which is not). Since the late 1980s, several small and one large-scale clinical trial, the Digitalis Investigation Group (DIG) study, have helped resolve many of the questions about the clinical efficacy of the digitalis glycosides. In the remainder of this chapter, I will refer to digoxin alone because this is the most widely used of the digitalis preparations.
Digoxin has well-documented inotropic effects (31,32) and it can block impulse transmission across the atrioven-tricular node. Atrial fibrillation occurs in up to 20% of patients with chronic heart failure, and the use of digoxin is probably least controversial in this setting. The fact that digoxin has only modest positive inotropic effects has been used to argue against its use in chronic heart failure patients who are in normal sinus rhythm. The rebuttal to this argument is that whatever inotropic effect is present, it is enough to produce clinical benefits. In fact, one could argue from the opposite perspective that more intense long-term inotropic support of the heart may actually have deleterious effects in patients with chronic heart failure (33,34). It appears, however, that the noncardiac effects of digoxin, such as enhancing baroreceptor sensitivity (which is diminished in chronic heart failure), are related to clinical efficacy. Digoxin inhibits peripheral sympathetic nervous system activity in patients with chronic heart failure (35). The effects on sympathetic nerve traffic appear to be due to an intrinsic property of the drug rather than to withdrawal of reflex sympathetic stimulation because a comparable improvement in cardiac performance with dopamine does not have the same effect on sympathetic activity. This, of course, raises the question of whether or not digoxin would still have beneficial effects if it were given on top of therapy, which already includes a β-blocking agent.
Although several small clinical trials published in the 1980s suggested that digoxin might be of value in patients with chronic heart failure who were in sinus rhythm (36,37), it was not until somewhat later that this point was
established. One of the first trials to demonstrate the value of digoxin compared its effects to those of captopril (an ACE inhibitor) and placebo (38) in patients who had mostly mild [New York Heart Association (NYHA) functional Class I and II symptoms] heart failure and were stable on diuretics alone (after digoxin was withdrawn). Compared with placebo- and captopril-treated patients, the digoxin-treated group demonstrated a significant increase in LV ejection fraction. The captopril-treated patients experienced a significant increase in treadmill exercise time and NYHA class compared with the placebo group. Despite the relatively stable condition of the patients on entry into the study, the brief duration of the trial, and small numbers involved, there was a significant reduction in chronic heart failure-related clinical events in both the captopril and digoxin groups. These results show that in patients with mild chronic heart failure symptoms, both digoxin and captopril are effective therapy and that diuretics alone are not sufficient.
established. One of the first trials to demonstrate the value of digoxin compared its effects to those of captopril (an ACE inhibitor) and placebo (38) in patients who had mostly mild [New York Heart Association (NYHA) functional Class I and II symptoms] heart failure and were stable on diuretics alone (after digoxin was withdrawn). Compared with placebo- and captopril-treated patients, the digoxin-treated group demonstrated a significant increase in LV ejection fraction. The captopril-treated patients experienced a significant increase in treadmill exercise time and NYHA class compared with the placebo group. Despite the relatively stable condition of the patients on entry into the study, the brief duration of the trial, and small numbers involved, there was a significant reduction in chronic heart failure-related clinical events in both the captopril and digoxin groups. These results show that in patients with mild chronic heart failure symptoms, both digoxin and captopril are effective therapy and that diuretics alone are not sufficient.
The RADIANCE trial evaluated the effects of digoxin withdrawal in 178 patients with NYHA Class I to III chronic heart failure (39). Patients were required to have an EF below 35%, to be in normal sinus rhythm, and to have been clinically stable on a regimen of digoxin, diuretics, and ACE inhibitors for a period of 8 weeks prior to randomization. During 12 weeks of follow-up, clinical deterioration occurred more frequently in patients who had digoxin withdrawn (28%) than in patients who had digoxin continued (6%). Digoxin withdrawal was associated with a significantly higher likelihood of an increase in diuretic dose, need for emergency room care, or hospitalization for chronic heart failure (25% versus 5% in the group that continued digoxin, p ≤ 0.001). Patients who had digoxin withdrawn also experienced a significant reduction in treadmill exercise time, distance covered during a 6-minute walk test, and in LV ejection fraction. A similar study conducted in patients who were not receiving ACE inhibitors reported comparable findings (40).
The effects of digoxin on mortality were evaluated in the large-scale DIG trial, in which 6,800 patients with mild to moderate heart failure were randomized to continue digoxin or have it withdrawn from their medical regimen (41). The results showed quite conclusively the absence of any survival benefit with digoxin. A 28% reduction in hospitalizations due to worsening heart failure (p <0.001) confirmed the beneficial effects with digoxin seen in smaller trials. The overall reduction in all-cause hospitalization was 6%. The DIG study also reported an insignificant but nonetheless disquieting increase in sudden death in patients receiving the drug.
The results of these clinical trials justify the use of digoxin in treating patients in sinus rhythm. They also provide evidence for guiding its administration. Digoxin is useful mainly for treating signs and symptoms of heart failure but it has no demonstrable effects on either the progression of LV dysfunction or on survival (41). Therefore, the use of digoxin should be reserved for those chronic heart failure patients who either require rate control of atrial fibrillation or those in sinus rhythm who remain symptomatic after optimization of other first-line therapies.
One of the advantages of digoxin is that it is relatively easy to use and can be given only once a day. Although the drug is not innocuous, the incidence of toxicity in clinical practice is certainly very far below the estimated 15% level that was reported in hospitalized patients in the early 1970s (42). This is likely due to multiple factors, including the avoidance of longer-acting digitalis glycosides, a higher degree of alertness to the signs and symptoms of digoxin toxicity on the part of practicing clinicians, increased recognition of the conditions in which digoxin toxicity is likely to occur, the availability of other agents to treat chronic heart failure both acutely and chronically (thus minimizing the need to “push dig” in patients who are doing poorly), greater attention to electrolyte abnormalities that could potentiate digoxin toxicity, the widespread availability of an assay to determine levels of digoxin in the blood, and general recognition that a low dose of digoxin is all that is necessary for treating heart failure patients (43).
The use of digoxin in the acute management of patients with decompensated chronic heart failure is much less common than in the past because other more potent inotropic and/or vasodilator agents that are rapidly acting and can be carefully titrated are now available. In stable patients, a maintenance dose of digoxin, ranging from 0.125 to 0.25 mg daily, is usually begun. A stable concentration in blood is usually obtained within 5 to 7 days and, when there are concerns about clearance, confirmed by measurement of the serum digoxin level. Continuous measurement of digoxin levels in stable patients is not needed. Even in situations where digoxin levels may be altered by concurrent therapy or worsening renal disease, most physicians simply reduce the dose of the drug. In some cases measuring digoxin levels is an important indicator of the overall compliance of the patient to the medical regimen and it can help sort out the cause of an episode of decompensation. Because there is little evidence that high-dose digoxin offers significant clinical benefit over low-dose therapy (44,45), the use of digoxin levels to titrate the dose of drug has little validity.
Angiotensin-Converting Enzyme Inhibitors
As mentioned in the section on pathophysiology and in other chapters of this text, activation of the RAS plays an important role in the pathogenesis of chronic heart failure. In general, elevations in plasma renin activity correlate with the severity of the condition (46). A reduction in cardiac output and renal perfusion appears to be a major factor in this process (46,47). Patients with asymptomatic LV dysfunction, however, may have increased levels of plasma renin activity, particularly if they are being treated with diuretic agents (28). The local, tissue-based RAS also appears to be upregulated in the setting of LV dysfunction and it appears to play a major role in the progression of the disease by promoting cardiac remodeling (15,16,48,49).
As summarized in Table 35-2, the adverse consequences of activation of the RAS provide a rationale for the use of ACE inhibitors in the treatment of chronic heart failure. The ACE inhibitors, however, may also have other actions that could account for their demonstrated clinical efficacy. Converting enzyme, in addition to blocking the breakdown of angiotensin I to angiotensin II, are also involved in the inactivation of bradykinin, substance P, and other
vasoactive compounds (50). The effects on bradykinin may be of particular importance because this substance can stimulate endothelial cells to release prostacyclin (51) and nitric oxide (NO) (52), both of which have vasodilatory properties. Thus, ACE inhibitor potentiation of bradykinin could promote vasodilation through pathways unrelated to the RAS (53). Bradykinin also may have antigrowth effects that could contribute to the effects of ACE inhibitors in preventing cardiac remodeling (54).
vasoactive compounds (50). The effects on bradykinin may be of particular importance because this substance can stimulate endothelial cells to release prostacyclin (51) and nitric oxide (NO) (52), both of which have vasodilatory properties. Thus, ACE inhibitor potentiation of bradykinin could promote vasodilation through pathways unrelated to the RAS (53). Bradykinin also may have antigrowth effects that could contribute to the effects of ACE inhibitors in preventing cardiac remodeling (54).
As a result of their ability to dilate peripheral vessels and to unload the heart, the ACE inhibitors have been shown to improve cardiac function (55,56,57). Initially, there is little impact on exercise capacity despite an increase in cardiac output and a reduction in ventricular filling pressures. However, over a period of weeks to months, exercise duration improves in patients with mild to moderate chronic heart failure (38,58,59). Symptomatic improvement also becomes manifest over time. The reasons for the temporal disparity between the acute cardiac and long-term clinical effects of the ACE inhibitors are uncertain. Enhanced peripheral vasodilation and tissue oxygen extraction during chronic (but not acute) therapy have been demonstrated (60,61). This could be related to alterations in tissue mechanisms for oxygen extraction, structural changes in blood vessels, or better conditioning (as cardiac function improves). ACE inhibitors also have been shown to enhance NO production in an experimental model of chronic heart failure (62), and this factor could account for the improved peripheral vasodilation and clinical benefits seen during long-term therapy.
One of the most important goals of the management of patients with chronic heart failure is to prevent further myocardial dysfunction. There is evidence that the LV undergoes progressive change in size, structure, and function over time following MI (7,8,9). Patients who survive an MI with more than mild myocardial damage experience progressive increases in chamber size and muscle mass (8,9). Both LV dilation and hypertrophy, unfortunately, are strongly related to both late deterioration in LV systolic function and to decreased survival (63,64,65,66,67). The fact that ACE inhibitors favorably affect both mechanical and neurohormonal factors that promote remodeling provided the rationale for evaluating their effects following MI. Initial studies demonstrated that ACE inhibitors prevented post-Mi increases in LV chamber size (10,68). SAVE and other well-designed, large-scale clinical trials which evaluated the effects of ACE inhibitors on the clinical course of MI survivors have demonstrated that they are associated with prolonged survival and a lower likelihood of developing heart failure or hospitalization (69,70,71,72,73,74,75)-effects that likely are associated with their ability to inhibit post-MI cardiac remodeling.
The effects of ACE inhibitors on survival in patients with LV dysfunction have been studied extensively (76,77,78,79). Some of the key features of trials evaluating ACE inhibitors are summarized in Table 35-3. Although ejection fraction cut-offs and other entry criteria varied somewhat between the trials, all patients had moderate to severe LV systolic dysfunction. Functional impairment ranged from the asymptomatic state to NYHA Class IV heart failure. The results provide conclusive evidence that ACE inhibitor therapy improves survival in a broad spectrum of patients with LV dysfunction (76,77,78,79). Moreover, they show that ACE inhibitors are effective in most subgroups of patients and that the benefits are not dependent on factors such as age, gender, etiology of chronic heart failure, serum sodium level, or treatment with β-blockers. ACE inhibitors appear to be more effective in reducing mortality in more severely ill patients (e.g., NYHA functional Class IV in CONSENSUS [74]) than in less symptomatic patients (e.g., NYHA functional Class I-II in the SOLVD Prevention arm [78]). There is also some evidence that efficacy of the ACE inhibitors is greater in those patients with more severe depression of LV ejection fraction (77,78) but that their beneficial effects may be attenuated when patients are receiving aspirin (80,81).
The effects of ACE inhibitors on cardiac remodeling in patients with LV dysfunction were evaluated in a subgroup of the SOLVD population (82). Although these patients had evidence of considerable LV dilation and hypertrophy at the time of randomization, patients assigned to placebo demonstrated progressive increases in LV volumes and muscle mass over the 12-month follow-up period, thereby indicating the relentless nature of cardiac remodeling. Further increases in LV volumes and mass, however, were prevented in the enalapril-treated patients. These results from a representative subgroup of the SOLVD study population suggest that the clinical benefits of the ACE inhibitors are related to their favorable effects on the remodeling process. They also show that the opportunity to prevent further increases in muscle mass and chamber volume even after considerable remodeling has occurred.
The side effects associated with ACE include hypotension, hyperkalemia, dizziness and syncope, taste disturbances, worsening renal function, cough, and angioneurotic edema. A reduction in blood pressure is expected with the initiation of therapy, and in most instances it is of little consequence. Patients at increased risk of developing clinically important hypotensive symptoms can be identified by the presence of advanced functional class (i.e., Class IV); recent aggressive therapy with diuretics; use of non-ACE inhibitor vasodilators; presence of low serum sodium levels (i.e., serum Na <130 mm L/L) (83); and borderline blood pressure at the time when therapy is initiated. Because many of these patients fall into categories that are most likely to benefit from ACE inhibitor therapy, the greater incidence of hypotension should not be considered a contradiction to initiating treatment. Experience from SOLVD and other trials demonstrates quite clearly that only a small number of patients with LV dysfunction could not be continued on ACE inhibitors because of hypotension or other side effects (74,75,76,77,78,79).
ACE inhibitor therapy should be initiated at a low dose which is uptitrated over a period of days to weeks depending on the clinical situation. Hospitalized patients in whom dose of drug can be increased in a controlled setting often reach the target dose within a matter of days, while uptitration of ACE inhibitors in the outpatient setting is usually more gradual. Patients should be educated about the possibility of hypotension, and a blood chemistry screen to assess renal function and serum potassium levels should be obtained at 1 week after initiation, following each increase in dose, and when the target dose has been
reached. Many candidates for ACE inhibitors have evidence of mild to moderate renal dysfunction prior to the initiation of therapy. A small bump in BUN or creatinine levels is common and should not cause undue concern. Sustained increases in BUN greater than 15 to 20 mg/dL or in creatinine greater than 0.2 to 0.5 mg/dL can often be successfully treated by adjusting therapy. In patients receiving diuretics or other non-ACE inhibitor vasodilators, a reduction in the dose of these drugs is often sufficient to correct the abnormality. If this tactic fails, one can consider reducing the dose of the ACE inhibitor. However, because the changes in BUN and creatinine are reversible, reduction in dose or discontinuation of ACE inhibitors should be undertaken only after careful consideration of the risk/benefit ratio of doing this.
reached. Many candidates for ACE inhibitors have evidence of mild to moderate renal dysfunction prior to the initiation of therapy. A small bump in BUN or creatinine levels is common and should not cause undue concern. Sustained increases in BUN greater than 15 to 20 mg/dL or in creatinine greater than 0.2 to 0.5 mg/dL can often be successfully treated by adjusting therapy. In patients receiving diuretics or other non-ACE inhibitor vasodilators, a reduction in the dose of these drugs is often sufficient to correct the abnormality. If this tactic fails, one can consider reducing the dose of the ACE inhibitor. However, because the changes in BUN and creatinine are reversible, reduction in dose or discontinuation of ACE inhibitors should be undertaken only after careful consideration of the risk/benefit ratio of doing this.
Table 35-3 Effects of Ace Inhibitor Therapy on Survival of Patients with Left Ventricular Dysfunction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Cough is a frequent complaint in patients with chronic heart failure (84). However, ACE inhibitor-related cough must be differentiated from cough due to a myriad of other causes (including worsening pulmonary congestion). The problem is highlighted by results from the treatment arm of the SOLVD study, where it was found that although 37% of patients treated with enalapril complained of cough, this symptom was also noted in 31% of placebo-treated patients (77). Thus, while cough due to ACE inhibitors is a real problem, its actual incidence is relatively low. A careful search for other causes of cough should always be undertaken before discontinuing ACE inhibitor therapy. There is no convincing evidence that one ACE inhibitor is more likely to provoke cough than another, and changing the ACE inhibitor is rarely successful in solving the problem. When cough does not resolve after the drug is stopped, one can conclude that the problem is not due to the ACE inhibitor, which should then be restarted while the real cause is sought.
There is a tendency for potassium levels to increase when ACE inhibitors are initiated. As evidenced in both arms of SOLVD (77,78), the change is small and of little or no clinical consequence in most instances. However, in patients with underlying renal dysfunction and in patients receiving potassium-sparing diuretics or large amounts of potassium supplementation, the increase in serum potassium level can on occasion be life-threatening. The condition can be treated by reducing either the non-ACE inhibitor factor (e.g., K+ supplement) or the ACE inhibitor itself. Vigilance on the part of the physician is necessary, however, to detect hyperkalemia.
An interesting but regrettable phenomenon that has developed with the use of the ACE inhibitors in patients with chronic heart failure is that they tend to be used at lower doses than were used in the studies that provided evidence of efficacy. The obvious reason for this strategy is the desire to avoid side effects. However, SOLVD and other large-scale clinical trials provide strong evidence that the ACE inhibitors tend to be well-tolerated (76,77,78,79). It is uncertain whether the same clinical benefits that were noted in the clinical trials will be present when the drugs are used at lower doses. In fact, the ATLAS study showed that high doses of ACE inhibitors are associated with greater improvement in the clinical course (85) and that they are well-tolerated (86). Thus, the use of target doses of ACE inhibitors (e.g., ramipril at 10 mg per day, enalapril or lisinopril at 20 to 40 mg per day, and captopril at 150 mg per day) is strongly recommended.
Angiotensin Receptor Blockers
Over the past 5 years angiotensin receptor blockers (ARBs) have gained an increasingly important role in the management of patients with heart failure. Their initiation into the treatment regimen was delayed largely because of the earlier availability of ACE inhibitors. However, ARBs are now recognized as being quite useful alternatives to ACE inhibitors in the management of patients with post-MI LV dysfunction and in patients with heart failure due to systolic dysfunction. There is also evidence that their addition to an ACE inhibitor can improve the clinical course of heart failure patients.
ARBs compete with angio-tensin II for occupancy of the type 1 angiotensin II receptor (AT1R) on cells throughout the body. Since most adverse cardiovascular effects of angiotensin II (such as salt and water retention, peripheral vasoconstriction, cardiac myo-cyte hypertrophy, and fibroblast activation) are mediated through the AT1R, blockade of this receptor should be protective. Moreover, ARBs block the effects of angiotensin II regardless of whether it is generated by the usual pathway involving ACE or through alternative pathways. In the heart an alternative pathway involving chymase that is released from mast cells is believed to be particularly important in the generation of angiotensin II (87). An added benefit of the ARBs may be related to so-called shunting of angiotensin II from the AT1R to type 2 angiotensin receptor (AT2R), which is believed to mediate antigrowth effects. There is also evidence that ARBs increase the expression of a homologue of ACE termed ACE2 (88). Activity of this enzyme may be cardioprotective in that it acts to break down angiotensin II while generating Ang-(1,2,3,4,5,6,7). This heptapeptide has been shown to have both vasodilatory, antiproliferative, and antigrowth properties that are mediated by activation of a novel non-AT1 or AT2 receptor (89,90,91).
The efficacy of ARBs in treating patients with post-MI LV dysfunction was demonstrated in the VALIANT study in which the effects of valsartan were compared to captopril (92). This particular ACE inhibitor was chosen as the active comparator in VALIANT based on its proven efficacy in treating patients with post-MI LV dysfunction in the SAVE study. An additional arm of the VALIANT study included patients treated with a combination of captopril and valsartan. The results demonstrated that valsartan was equally as effective as captopril in reducing cardiovascular morbidity and mortality but that the combination was associated with increased adverse events without improving outcomes. In VALIANT (as in previous studies), baseline echocardiographic parameters were shown to be powerful predictors of all major outcomes. Treatment with captopril, valsartan, or the combination of captopril plus valsartan resulted in similar changes in cardiac volume, EF, and infarct segment length between baseline and 20 months after MI, indicating relative equivalence in protectio against adverse cardiac remodeling. Based on these results
the use of an ARB has been accepted as an alternative to an ACE inhibitor in the treatment of patients with post-MI LV dysfunction.
the use of an ARB has been accepted as an alternative to an ACE inhibitor in the treatment of patients with post-MI LV dysfunction.
The effects of an ARB in ACE inhibitor-intolerant patients were assessed in the CHARM-Alternative study (93). This trial included more than 2,000 ACE inhibitor-intolerant patients (due to cough in 72%, symptomatic hypotension in 13%, and renal dysfunction in 12%) with symptomatic heart failure due to LV systolic dysfunction who were randomized to receive treatment with either candesartan or placebo. The primary outcome was the composite of cardiovascular death or hospital admission for heart failure. As shown in Figure 35-2 during a median follow-up of 33.7 months, candesartan was associated with a 23% risk reduction for the combined endpoint (p = 0.0004). Each component of the primary outcome was reduced, as was the total number of hospital admissions for CHF. Study-drug discontinuation rates were similar in the candesartan (30%) and placebo (29%) groups. These results were further substantiated by a prior analysis of a group of patients in the Val-HeFT study who were not receiving an ACE inhibitor and in whom valsartan significantly reduced mortality (94).
The results of the CHARM-Alternative and Val-HeFT subgroup analysis provide persuasive evidence of the beneficial effects of ARBs as a treatment for heart failure due to systolic dysfunction in ACE inhibitor-intolerant patients. Previous comparison between the two approaches in the ELITE-2 study, however, indicated that the use of an ARB did not offer benefits that were superior to those of an ACE inhibitor (95). Thus, ARBs remain an alternative for use in heart failure patients who are ACE-intolerant. They could also be continued if the patient was receiving an ARB for a pre-exisiting condition such as hypertension at the time that he or she developed heart failure.
The fact that ACE inhibitors and ARBs have distinct phar-macologic profiles raises the possibility that their combined effects have incremental benefits in heart failure patients. This hypothesis was tested in two large-scale trials, Val-HeFT and CHARM-Added. In Val-HeFT, valsartan was added to standard therapy, which in most cases included an ACE inhibitor. The results demonstrated a significant 13% reduction in the combined primary endpoint of morbidity and mortality (96). This reduction was due largely to a reduction in heart failure hospitalizations and there was no significant impact on mortality. Post hoc analysis of the Val-HeFT database also raised the possibility that the addition of the ARB to patients already receiving an ACE inhibitor and β-blocker might be associated with an increase in mortality. In CHARM-Added, 2,548 patients with NYHA functional Class II-IV CHF and LVEF 40% or lower who were being treated with ACE inhibitors were randomly assigned to either candesartan or placebo (97). After 41 months of follow-up, the composite of cardiovascular death or hospital admission for heart failure was reduced by 15% (p = 0.011). Candesartan reduced each of the components of the primary outcome significantly, as well as the total number of hospital admissions for heart failure. Of the patients enrolled in CHARM-Added, 55% were receiving a β-blocker at the time of randomization. Analysis of outcomes in this predefined subgroup failed to detect any evidence of a negative impact of adding the ARB to existing therapy, suggesting that the adverse interaction seen in Val-HeFT was likely a statistical artifact. Based on the results of these studies, the possibility of adding an ARB to standard therapy (including an ACE inhibitor and β-blocker) in heart failure patients who remain symptomatic despite treatment with an ACE inhibitor and β-blocker should be considered.
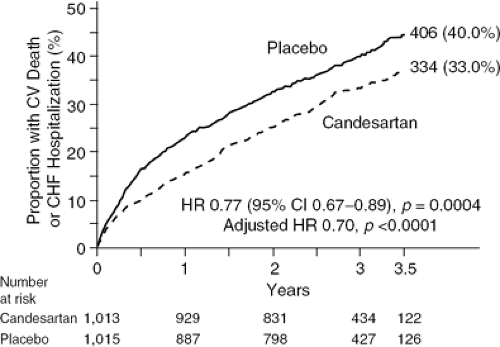 Figure 35-2 Results from the CHARM-Added study demonstrating a reduction in the primary end-point of cardiovascular death or heart failure hospitalization. (From Granger CB, McMurray JJV, Yusuf S, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003; 362:772–776 .) |
The side effects of the ARBs are similar to those of the ACE inhibitors except for the absence of cough and a much lower likelihood of angioneurotic edema. The recommendations for uptitration and dosing of the ARBs and for following laboratory assessment of renal function and serum electrolytes are the same as those given for the ACE inhibitors in the previous section. The likelihood of worsening renal function, hyperkalemia, and hypotension is significantly greater with the combination of an ACE inhibitor and ARB than with either drug alone. Vigilance is required when the combination is used in order to avoid untoward consequences.
β-Adrenergic Receptor Antagonists
Activation of the sympathetic nervous system is one of the most important factors responsible for the progression of heart failure. Sympathetic activation occurs early in the course (28) and it provides circulatory support by increasing heart rate and contractility and by constricting peripheral blood vessels (Table 35-2). Levels of circulating cate-cholamines increase in patients with chronic heart failure in proportion to the severity of the disease (28). Nor-epinephrine has a number of deleterious effects on the adult mammalian cardiomyocyte (98). Catecholamines promote myocyte growth, increase cell death by inducing both necrosis and apoptosis, desensitize β-adrenergic receptors, cause peripheral vasoconstriction, enhance arrhythmias, augment renin release, and impair sodium excretion from the kidney (99,100,101,102,103,104,105,106). Not surprisingly, there is a strong association between high sympathetic nervous activity and an unfavorable prognosis in patients with chronic heart failure (107).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


