Definition
Notes
Molecular or morphological criteria lo define dead cells
Loss of plasma membrane integrity
Plasma membrane has broken down, resulting in the loss of cell’s identity
(IF) Microscopy and/or FACS to assess the exclusion of vital dyes, in vitro
Cell fragmentation
The cell (including its nucleus) has undergone complete fragmentation into discrete bodies (usually referred to as apoptotic bodies)
(IF) microscopy
FACS quantification of hypodiploid events (sub-G1 peak)
Engulfment by adjacent cells
The corpse or its fragments have been phagocytosed by neighboring cells
(IF) microscopy
FACS colocalization studies
Proposed points-of-no return to define dying cells
Massive activation of caspases
Caspases execute the classic apoptotic program, yet in several instances, caspase- independent death occurs. Moreover, caspases are involved in non-lethal processes including differentiation and activation of cells
Immunoblotting
FACS quantification by means of fluorogenic substrates or specific antibodies
ΔΨm dissipation
Protracted ΔΨm loss usually precedes MMP and cell death; however, transient dissipation is not always a lethal event
FACS quantification with ΔΨm, -sensitive probes Calcein- cobalt technique
MMP
Complete MMP results in the liberation of lethal catabolic enzymes or activators of such enzymes. Nonetheless, partial permeabilization may not necessarily lead to cell death
IF colocalization studies
Immunoblotting after subcellular fractionation
PS exposure
PS exposure on the outer leaflet of the plasma membrane often is an early event of apoptosis, but may be reversible. PS exposure occurs also in T-cell activation, without cell death
FACS quantification of Annexin V binding
Operative definition of cell death, in particular in cancer research
Loss of clonogenic survival
This method does not distinguish cell death from long-lasting or irreversible cell cycle arrest
Clonogenic assays
The term CD includes three modalities leading to this result. Some atypical forms will be described at the end of this chapter.
15.2 Apoptosis
Here it must be reminded, as Whelan et al. [3] and other authors point out, that apoptotic programmed CD in the nematode Caenorhabditis elegans has been conserved over 600 million years during the development of this organism.
Moreover, since it involves the Mitochondria (MITO) it gives a significant insight into the function of these important organelles, as will be further described.
Its name derives from the Greek, meaning falling (ptosis)-off (apo). While necrosis (N) is characterized by cell swelling due to plasma membrane rupture, this membrane in Apoptosis (APO) remains intact (not to be confused with the rupture of the outer mitochondrial membrane) while the cell shrinks, together with the nucleus. Morphologic characteristics of the nucleus in APO are chromatin condensation, and nuclear fragmentation. In a further stage, the nucleus break-up is also called karyorrhexis (rhexis = rupture).
The cells emit processes or pseudopodia (budding) which contain pyknotic nuclear fragments, as described 20 years ago by Majno and Joris [4] who mention that the first microscopic picture of APO probably appeared in 1886. These “buds” are phagocytosed by local resident cells, usually without causing an inflammatory response.
Apoptotic bodies contain fragments of both cytoplasm and nucleus.
Whelan et al. [3] and Crow et al. [6] from the group of Richard Kitsis describe the two pathways which mediate cell death in APO and which are intimately entwined:
(a)
Extrinsic pathway or death receptor (DR) pathway [7]. This is effected by the binding of death ligands, Fas and TNF-α being prime examples. Fas binds to the cell surface receptors, binding to the adaptor protein FADD (Fas- associated via death domain). FADD recruits procaspases-8 and-10 through the DISC (multiprotein death inducing complex) which activates procaspase-3 and Bid, (BH3 – interacting domain death agonist) which activates pro-to caspase-3.
(b)
The intrinsic pathway or canonical or mitochondrial pathway. It also involves the endoplasmic reticulum (ER). This combines extra-cellular and intra-cellular stresses of any type; however in the former, deficiency of nutrients, radiation, drugs and physical stress predominate, while in the latter the main factors are oxidative stress, DNA damage and protein misfolding. Both types of signals are transmitted to the MITO by the BH3 only proteins which will be further described. These stimulate the release of cytochrome-c and other apoptogens as described by Baines [7].
Apoptotic stimuli are ultimately mediated through the proapoptotic Bcl-2 protein family. This family is differentiated as follows:
1.
The antiapoptotic members, of which Bcl-2 (B cell Leukemia/lymphoma 2) and Bcl-XL (long isoform) are the representatives.
2.
The proapoptotic members, which include many proteins:
Main players are (Bax (Bcl-2 associated X protein), Bak (Bcl-2- homologous antagonist killer and BH3-only (Bcl-2 homology domain 3-only) proapoptotic proteins: Bid (BH3 interacting domain death agonist), Bad (Bcl-2-antagonist of cell death). Bim (Bcl-2-interacting mediator of cell death). Bmf (Bcl-2 modifying factor), Noxa, Smac/DIABLO which will be discussed further on, Puma (p 53 upregulated modulator of APO), which is essential for DNA damage-induced APO, BNIP3 (Bcl-2 and 19 kDa interacting protein-3). Either Bax or Bak are necessary for this process; they are translated to the outer mitochondrial membrane (MOM). They stimulate the release of cytochrome c and other apoptogenic mitochondrial proteins into the cytosol. Bax activation may be mediated through calpain; as this protein will also be mentioned as an effector of N, its participation in both processes may represent another link in the crosstalk of the two processes. Bax and Bak, which are located at the ER as well as the MITO increase ER Ca+2 stores, while Bcl-2 decreases them. The increased ER Ca2+ increases the release of Ca2+ in the cytoplasm, causing APO [6].
Cytochrome c and other apoptogens (importantly procaspase-9) are gathered in the apoptosome, activating pro-caspase 9, which activates caspase-3, the main mediator of apoptotic CD. Many other effectors are involved in this process, as described by Whelan et al. [3] and Crow et al. [6].
According to Whelan et al. [3] the main inhibitor of APO in the intrinsic pathway are the Bcl-2 proteins. However, the IAP proteins (inhibitor of APO family) are also important, since they inhibit activated caspase-3 and −7.
Other APO inhibitors are: FLIP (FLICE-like Fas associated death domain protein-like-interleukin-1-converting enzyme like inhibitory protein), and ARC (apoptosis repressor with a CARD).
Apart from their role in APO, several BH3 only members also regulate cell-cycle, DNA repair and metabolism, but are also involved in the crosstalk of APO with the third cell death process to be discussed next, autophagy (AUTO) by liberating Beclin 1 from its binding within the Bcl-2/Bcl-XL complexes.
Crompton [8] underlines the multifaceted actions of the Bcl-2 family proteins, with the Bcl-2/Bax ratio determining resistance to APO. This ratio is very widely used in REM myocardium studies.
The JNKs (superfamily of MAP-kinases) are instrumental in both cell proliferation and APO; the balance can be tilted by many factors.
Dhanasekaran and Reddy [9] point out that JNK signals APO by various mechanisms: It increases expression of TNF-α, Fas and Bak. It also phosphorylates the p53 family of proteins. It is also critical for the release of cytochrome c, and induces cleavage of Bid; it promotes the release of Smac/DIABLO, which antagonizes cytosolic IAPs, from the inter-membrane space to the cytosol, stimulating caspase activation.
The mitochondria production of APO is to a large extent mediated by the MITO. A description of their structure and function is considered helpful.
They are located into the intermyofibrillar spaces, underneath the sarcolemma. Their location permits more efficient ATP supply to the myofibrils.
The MITO drive two different CD mechanisms (APO and N) through the mitochondrial permeability transition pore (MPTP). They are surrounded by two membranes [7, 8, 10], with a narrow intermembrane space between them.
15.2.1 The Outer Membrane (MOM or OMM)
It has many pores based on protein, which allow passage of ions and molecules. It drives APO.
This is the action site of the already mentioned pro-death members of the Bcl-2 family. Under the response to noxious stimuli, Bax which normally resides in the cytosol, is translocated to MOM and the endoplasmic reticulum (ER).
Thus it causes MOM permeabilization and the release of pro-apoptotic proteins from the inter-membrane space into the cytosol, such as cytochromes, Smac/DIABLO and endonuclease-G (endo G). Cytochrome c binds to the cytosolic protein apaf 1 and thus causes the formation of the “apoptosome” which activates the caspase-9 and −3 system [7].
15.2.2 The Inner Membrane (MIM or IMM)
This has more restricted permeability, much like the cell plasma membrane. It is involved in electron transport and ATP synthesis, and is spanned by the Mitochondrial Permeability Transition Pore (MPTP) which drives N.
This pore is inhibited by low pH <7:0 thus it is closed during ischemia, which corresponds clinically to persistent artery occlusion. When reperfusion occurs, there is restitution of pH and acceleration of cytosolic Ca2+ increase (which is slow during ischemia). The cell deals with this explosive Ca2+ increase by taking it up into the mitochondria via the MITO Ca2+ uniporter a protein that uses the negative ΔΨm to drive Ca2+ into the matrix [12].
There exist many MPTP activators, such as Ca2+, ROS, Pi inorganic phosphate which uses after adenine nucleotides are depleted after the onset of ischemia and a reduction of the ATP/ADP ratio. From the pharmacological agents cyclosporine-A is a typical inhibitor; its beneficial effects on I/R injury [8] will be discussed later.
The MPTP is very well described by Baines [7], Crompton [8], Lemasters et al. [10], and Weiss et al. [11]: They state that the MITO use electron transport to develop an electrochemical gradient across the MIM (membrane space and matrix). This is formed by the MIM membrane potential (ΔΨm or ΔCm according to Weiss et al. [11] ≈ −200 mV), and a proton gradient (ΔpH). The electrochemical gradient is employed by ATP synthase to phosphorylate ADP to ATP. When MPTP opens, ΔΨm depolarizes, and the synthase actually starts consuming ATP in a (futile) attempt the restore the proton gradient; solutes up to 15 kDa pass the MIM, and cause MITO swelling. As MITO swell, the MIM stretches, but actually it is the MOM which ruptures: The surface of MIM is enhanced by the cristae, parallel invaginating membrane structures. The MOM rupture effects the release of pro-apoptotic proteins from the inter-membrane space, notably cytochrome c, Smac/DIABLO and endonuclease – G (endoG). Cytochrome c binds to the cytosolic protein apaf1, creating the “apoptosome” which activates the Caspase-9 and 3 protease system, while EndoG translates to the nucleus, mediating DNA fragmentation, as described by Baines [7], who also points out that the Bcl-2 proteins permeabilize the MOM while ROS and Ca2+ the MIM. The role of ROS production is complex. MITO ROS can trigger MPTP opening, with further ROS release creating a cascading process known as ROS-induced ROS release mechanism.
Some further molecular characteristics of the MPTP should be given:
(a)
The voltage-dependent anion channel (VDAC) resides in the MOM. This protein facilitates efficient transport of ATP/ADP across MOM; According to Baines [7], this protein, although very abundant, is not an essential component of the MPTP.
(b)
The Adenine nucleotide translocase (ANT) family resides in the MIM and facilitates the exchange of ATP and ADP across it. It is considered an essential factor, although recently some doubts have emerged about its central role. Crompton [9] points out that VDAC and ANT can interact strongly in junctional sites between the two membranes. They form a complex which can recruit additional proteins. One of these is Cyclophilin-D (CYPD).
15.2.3 Cyclophilin-D
This protein resides in the MITO matrix. Its activation opens the MPTP, while its inhibition (by cyclosporine-A or sangliferin A) is cardioprotective [12]. Interestingly according to Lemasters et al. [10] CsA prevents both N and APO.
The MITO phosphate carrier (PiC) is a CypD-interacting protein which regulates ATP synthesis. Its overexpression induces APO.
Apart from the MPTP, another death channel has been described, the mitochondrial apoptosis channel, which is regulated by the Bcl-2 protein family [12]: The Mitochondrial calcium uniporter (MCU), through which. normally, Ca2+ enter the MITO. As Pan et al. [13] describe, under normal conditions entry of Ca2+ in the MITO augments their ATP production to match its demand up to 10–20 fold. However, larger increases can induce CD. Normally, most unstimulated cells maintain Ca2+ in the range of 100–200 nM in the cytosol and the MITO through the function of pumps in the plasma membrane and the ER.
The MCU blockade can prevent abnormal increase Ca2+ (and Fe2+ entry).
Thus it is suggested that MCU is required to cope with increased Ca2+ cycling.
During ischemia, the closed state of the MPTP, effected by a decrease in pH, activation of Pi, and a mild increase of Ca2+ and ROS results into limited damage. At reperfusion, the opening of the MPTP is characterized by an increase of the pH, the Pi, the Ca2+ and an explosive increase of ROS resulting into irreversible damages.
Lemasters et al. [10] recapitulate that the MPTP is involved in both APO and N.
Another question which has been advanced is whether APO can be induced without the all-important caspase-3 activation [3, 12].
Studies in mice and C. elegans provide evidence for caspase independent CD. The apoptosis-inducing factor (AIF) is such a candidate.
Susin et al. [14] have shown that AIF can cause a picture of mitochondrial- effected APO which cannot be prevented by a wide ranging caspase inhibitor. It does not need Apaf 1 or procaspase-9 for its action. However, AIF is required for cell death mediated by poly-ADP ribose polymerase (PARP), which is a main component of N, as will be further described. It also interacts with cyclophilin A, and triggers the release of cytochrome c. They suggest that it has a direct effort on isolated nuclei, effecting chromatin condensation and fragmentation which can be caspase-independent. They also postulate that BH3-domain only protein may act independently of Apaf-1 and caspases.
15.2.4 Other Pathways Involved in APO
15.2.4.1 The Tumor Suppressor p53
It exerts its pro-apoptotic action through activation of multiple pro-apoptotic genes, i.e. Bax, Bid, Puma, apaf-1, caspase 6 and Fas.
The endoplasmic (or sarcoplasmic in muscle cells) reticulum pathway ER.
It causes an increase in intracellular Ca2+; its stores are increased by Bax and Bak which-apart from the outer mitochondrial membrane –are also located in the ER, together with the antiapoptotic Bcl-2 which has an opposing effect.
The HSP-70 which a well known cardioprotective chaperone reduces ER stress. It also reduces the apoptosome formation and AIF activation.
15.3 Necrosis
Necrosis (N) to the non-researcher has been considered the prevalent form of CD. It derives from the Greek νεκρός = corpse, dead person) It is usually considered to be non-programmed, in contrast to the two forms of Programmed Cell Death (PCD), Apoptosis and Autophagy.
However it will be subsequently shown that this distinction is not absolute.
Thus, some authors are bringing back the originally employed term oncosis, which is a predominantly morphologic term [6, 15]. The morphologic differences of the two modalities are well presented by Feuerstein et al. [15] (Fig. 15.1).
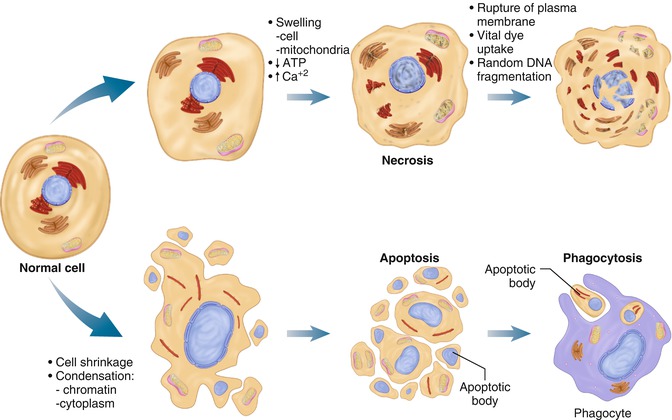
Fig. 15.1
Morphological differentiation of necrosis from apoptosis. This figure illustrates cardinal features of the histological appearances of cell death by either necrosis or apoptosis. The integrity of the plasma membrane and cell swelling are marked in necrosis, whereas cell shrinkage and fragmentation are most common in apoptosis. Most notable is the discrete process of phagocytosis of the “apoptotic bodies” by resident cells eliminating apoptosis, whereas cell eruption and content leakage (events triggering inflammation) are the final outcome of necrosis (Reproduced from Feuerstein et al. [15], copyright 1997 with permission from Elsevier)
The main causes of N are: The impact of a stress or insult beyond the protective capacity of the cell, which causes rupture of the plasma membrane and swelling of the MITO and endoplasmic reticulum (this is why the term oncosis has also been used. It is accompanied by nuclear pyknosis. However, the nuclear chromatin condensation is not as intense as seen in APO (Fig. 15.1).
The increased membrane permeability and rupture has two results: cellular and extracellular.
1.
Cellular
The cell and its organelles swell, as already described.
2.
Extracellular
Necrotic cells release their intracellular content, thus they induce inflammatory reactions, both of adjacent cells but also systemically. This causes activation of innate immunity, as will be further described.
N occurs because ATP cellular supply is exhausted. In what sequence this is associated with membrane rupture is difficult to ascertain, as will be further discussed. The decline in function of the ATP-dependent ion pumps in the membrane lead to the opening of the “death channel” which results in colloid osmotic forces and entry of cations inside the membrane,
The “death channel” is selectively permeable to cations and inhibitable by glycine [16].
Hotchkiss et al. [12] very pertinently point out that acute and profound decreases in ATP induce N, while modest yet chronic decreases of ATP induce APO.
Various types of insults have been described: Loss of blood flow is what mostly concerns the cardiologist, but physical trauma, heat, and radiation are also significant factors.
In what instances is N seen in the heart?
In the cardiovascular context, it is mainly caused by the abrupt interruption of blood flow, especially during coronary occlusion in the course of an acute myocardial infarction (AMI). The emergence of APO was widely identified when reperfusion was introduced in the therapy of MI.
Baines [7] reviews work showing that the MPTP is inhibited by low pH, and that it is quiescent during ischemia (occlusion).
However N is also seen as a result of combination of ischemic and reperfusion injury.
Early on it must be stressed that the limits between N and APO are becoming less distinct. Thus, very experienced workers refer to cell death as occurring through “necrosis or apoptosis” in I/R [17].
Also, Baines [7] points out that if a stress is severe and/or prolonged ATP (which is required for APO) will be significantly depleted and N will occur.
It is proposed that the hypercontraction caused by I/R in early experiments is a characteristic component of contraction band necrosis [3].
It is postulated that Ca2+ overload at higher magnitudes can cause N and at lower APO. Garcia-Dorado et al. [18] believe that APO is not a main component of Ca2+ overload –induced. CD. Due to the increase of membrane permeability the necrotic cells swell (while they characteristically shrink in APO). The mitochondria also swell. As already stated cellular contents during N spill into the extracellular space (and can be detected the periphery-plasma) inducing an inflammatory response [13]. This is mostly ascribed to molecules such as high mobility group protein B1 (HMGB 1).
Zong and Thompson [19] point out that N may enter the fate cycle of a cell and that the inflammation that it elicits may be a warning response. Thus, according to Whelan et al. [3] some authors have used the term Necroptosis, which will be discussed later on.
Galluzzi et al. [1], Whelan et al. [3] and Hotchkiss et al. [12] stress an intriguing point: That N may be less accidental and more regulated than originally thought.
Thus Galluzzi et al. [1] postulate that there exists a stereotyped cascade of events in the various models of N. Another aspect which they refer to is that N can occur during mammalian development, i.e. the death of chondrocytes during bone growth, or in the adult the loss of intestinal epithelial cells.
Luke et al. [20] have described that the intracellular serpin-6 which “regulates” N, exibits a prosurvival function by blocking N, protecting against hypotonic shock, heat shock, oxidative stress, and cation channel hypertrophy, thus casting doubt on its completely accidental nature.
15.3.1 Necrotic Pathways
According to Whelan et al. [3] N can be signaled by two pathways:
1.
The first involves death receptors, such as Tumor Necrosis Factor-α receptor 1 (TMFR1). This receptor can promote survival, APO or N. Laster et al. [21] stress the complex nature of the cytotoxic response that it can induce: In the rat fibroblast F17 cell line it induced typical APO while in the L-M line it caused N. Further details are beyond the scope of this review. However, one interesting aspect must be mentioned, the role of procaspase-8, which when activated can induce APO, but when inhibited, the result is N, through an increase of ROS production.
2.
The second necrotic mechanism is effected by the mitochondrial permeability transition pore (MPTP) which as already stated is situated in the inner mitochondrial membrane, actually on its inner leaf facing the intermembrane space.
This process is regulated by Cyclophilin D. MPTP can be opened by increases in oxidative stress, increased phosphate concentration, adenine nucleotide depletion, and importantly an increase of the matrix Ca2+.
Its opening causes the break-down of intracellular ATP.
Cyclophilin-D regulates MPTP pore opening and swelling of the mitochondria hallmark of N and cell death.
Cytoplasmic Ca2+ through MPTP opening can lead to N, in the worm and in the mammal through the proteases calpains, which are activated by Ca2+, and cathepsins which are released from the lysosomes during stress.
According to Zong and Thompson [19] calpain activation leads to N through cleavage of the Na+/Ca2+ exchanger in the plasma membrane and a sustained secondary intracellular Ca2+ overload.
However, according to Whelan et al. [3] calpain can also induce APO through Bax and Bid, and autophagy, through cleavage of Atg 5 (autophagy related 5 homolog) which will be described later. According to the same authors, the lysosomal cathepsins leak out explosively of the lysosomes, digest molecules and lead to N; under less abrupt conditions; partial lysosomal rupture leads to APO. They point out that N, APO and AUTO can be initiated by the lysosomes, whose integrity is preserved by HSP70.
Syntichaki et al. [22] point out that they are the result of perturbation of intercellular Ca2+.
Hotchkiss et al. [12] delineate the following mediators of N: ROS, Ca2+-activated non-lysosomal proteases calpains and cathepsins. These proteases are activated by oxidative stress and Ca2+ overload. Prolonged elevation of catecholamines Ang II and other vasoactives hormones may well be involved.
Whelan et al. [3] describe how ischemia-induced hypoxia increases intracellular acidosis, which extrudes H+ from the cell by Na+/H+ exchange leading to an increase of intracellular Na+, which through the Na+/Ca2+ exchanger leads to an intercellular Ca2+ increase, with the cell swelling effects which have already been described.
At reperfusion, Ca2+ induced Ca2+ release is effected through the ER(SR), or sarcoplasmic reticulum and MPTP opening is induced in some forms of N, such as DNA damage from alkylating agents mainly through ATP depletion. They also describe that HMGβ1, which as already stated is released as an inflammatory elicitor during N, can provide a switch from APO to N.
Opening of the MPTP pore is a major component of necrotic cell death. At R, APO cell death occurs at a lower incidence than N, but for a more extended time: APO occurs as early as 30 min and as late as 3 days after R initiation [23].
According to Whelan et al. [3], Poly-ADP-ribose-polymerase (PARP) is a main player in N occurring through DNA damage from alkylating agents.
15.4 Autophagy
This term derived from the Greek (auto = self and phagy = eat), delineates a process of death triggered by a sub-lethal stress involving nutrient deprivation. This causes the cell to recycle its own non-essential redundant or damaged organelles. Kroemer et al. [2] point out that probably one should define CD not occurring through, but with AUTO. The same authors point out that cells with the characteristics of autophagic CD can still recover when the noxious stimulus is withdrawn.
According to Clarke et al. [24], AUTO occurs regularly during the development of an organism, as in the amphibian tail during metamorphosis.
Three forms have been described: Macroautophagy will be further discussed. Chaperone mediated AUTO refers to a process mediated through chaperone proteins, such as HSC70 and Heart Shock Protein (HSP)70; these bind selectively to the substrate and deliver it to the lysosomes for degradation. In micro-autophagy, the material to be phagocytized is surrounded by invagination of the lysosomal membrane. From this point on, macro-autophagy will be described, in which the material to be phagocytized is sequestered within the characteristic body, the “autophagosome” contained by a double membrane vesicle.
This body can be well seen with electron microscopy. Its formation is governed by autophagy-regulating (atg) proteins, one of which interestingly a hallmark of AUTO-is Beclin-1(atg 6) [25]. Characteristically Beclin-1 is a BH3- only protein already described to be active in APO. Moreover, the anti-apoptotic proteins Bcl-2 and Bcl-XL can also inhibit starvation-induced AUTO.
Thus Bcl-2 plays a dual role: it can be pre-survival in the MITO, by blocking APO, and detrimental in the ER, by inhibiting AUTO.
Additional factors are the phosphatidylinosital-3-kinase (PI-3 K) and the mammalian target of rapamycin (mTOR) which inhibits autophagosome formation.
Whelan et al. [3] point out that under energy availability (fed condition) mTOR inhibits AUTO by phosphorylating and activating Atg proteins.
However, under starvation, reduced PI3K-Akt signaling decreases mTOR activity and activates AUTO.
After inclusion in the autophagosome, its components are lysed by the action of the lysosomes, providing the cell with amino acids, free fatty acids and energy.
Nucleus pyknosis can be seen, but is not so prominent as in APO. Thus, nucleus pyknosis is a characteristic of all three death modalities. Autophagosomes fuse with the lysosomes, the enclosed cytoplasmic material is degraded by acid hydrolysis, and the nutrients are released to the cytosol for recycling
Wang et al. [26] describe the multiple ways in which AUTO participates in metabolic remodeling: It provides amino acids, nucleotides, sugars and lipids. The former enter the tricarboxylic (TCA) acid cycle. Nucleotides can be recycled into new nucleic acids or enter the pentose phosphate pathway (PPP). Sugars can participate into new glucose synthesis and glycolysis, while lipids undergo β-oxidation. Together with energy production, the above processes contribute to new membrane generation biosynthesis. Starvation nutrient deprivation, hypoxia, ROS, damaged organelles and protein aggregates can induce AUTO, in an mTOR dependent process.
According to Dhesi et al. [27], the process of AUTO involves five steps:
1.
Formation of the isolation membrane or phagophore.
2.
Expansion of the phagophore.
3.
Formation of the double membrane.
4.
Fusion of the autophagosome with a lysosome to form the autolysosome.
5.
Digestion of the content together with the inner membrane layer by the lysosome hydrolases.
However, there is some discussion if these processes can actually cause CD, converting AUTO from a survival to a death process, if it is overexpressed. The increased number of autophagosomes in dying cells might signify that they are associated with CD, but it can well be a sign of failure of the cell to survive despite sacrificing its vital components, according to Hotchkiss et al. [12].
Thus, Dhesi et al. [27] believe that it is a beneficial process under most conditions. They also describe ER-phagy, or reticulo-phagy which selectively targets the ER, which actually forms the dual membranes described.
Gottlieb and Mentzer [28] also point out that it is a beneficial process under most conditions. Notably they enumerate many cardioprotective agents which induce AUTO (diazoxide, ranolazine, rapamycin, statins). They describe an additional term, mitophagy which is a specialized form which causes elimination of the MITO, which have been reported to be contained within the autophagosomes. Thus, they could be protected from “a mitochondrial stampede” causing widespread cell death by the “ROS begetting ROS production” process already described. They further stress that this may be the more important mechanism, since the amount of energy production may not be critical. They further enlarge upon the adaptive versus maladaptive instances, some of which will be further described. They conclude that the bulk of available evidence supports the protective role of AUTO.
Another question has arisen whether AUTO can be an adaptive response in desmin-related cardiomyopathy which is characterized by overexpression of mutated ab crystalin (cry ABR120G). It is being discussed by two investigative groups.
Tannous et al. [29] believe that AUTO increases by twofold in order to cope with the increase of misfolded protein aggregates seen in this disorder. Blunting AUTO dramatically hastened progression to heart failure.
Maloyan et al. [30] remark that mice with this cardiomyopathy show progressive MITO abnormalities and activation of CD through APO. By preventing APO through crossing this mice with transgenic stains with cardiac-specific Bcl-2 overexpression, they found decreased MITO abnormalities, attenuation of APO, reduction of protein aggregation, restitution of cardiac function, prevention of cardiac hypertrophy and a prolongation of survival by 20 %. However, the animals still died prematurely, thus inhibition of APO resulted in AUTO upregulation and increased N according to the authors, as detected by an increase of Evan Blue Dye cells, a technique which detects necrotic CD-related abnormalities. It is not clear from their work if CD is overall reduced or not.
Zhu et al. [31] have a different opinion as regards hemodynamic stress and AUTO: They consider that AUTO increases after transverse aortic constriction representing a maladaptive process. These questions will be further addressed when the three forms of death are assessed in the principal cardiological conditions.
Ferdous et al. [32] provide a very comprehensive review. They differentiate among three forms of AUTO activity: Basal, which is adaptive. This includes homeostatic protein control, ROS detoxification and removal of defective MITO. Maladaptive, which consists of excessive catabolism, MITO elimination, adverse REM, and reduced cell survival, or complete auto absence.
These authors point out the role of FoxO (Forehand box-containing protein, O subfamily) in governing AUTO.
Cao et al. [33] describe the role of this protein, which is activated by mechanical unloading and acts through the AUTO-lysosomal and ubiquination-proteasomal pathways to produce cardiac atrophy. Interestingly, it triggers BNIP-3 (Bcl-2 and 19-kDa interacting protein 3) which is also involved in APO.
15.4.1 The Role of the Ubiquitin Proteasome System (UPS)
It is often mentioned, but its actual role has not been adequately defined. Calise and Powell [34] provide a recent inclusive review.
This system, in parallel with the various mechanisms involved in CD has definite roles in cell-cycle control, protein turnover and quality control, cell signaling and APO.
According to the authors, the system is closely involved in myocardial ischemia. Its four components include the proteasome, ubiquitin (UB), the ubiquitination machinery and the deubiquitinases. It is an energy-requiring (ATP) process.
The proteasome can be involved in the removal and clearing of oxidized and damaged proteins. It can also form a hybrid, the “immunoproteasome”, since the ubiquitin system is also involved in immune response and antigen presentation.
Ubiquitin (Ub) is involved in protein degradation.
The UPS and specifically the proteasome, becomes dysfunctional in ischemia, both myocardial and cerebral. Its ATP-dependent activity is mainly affected, decreasing proteasome activity. It is also affected by oxidative damage. As in so many aspects of CD, the authors describe that whether a proteasome inhibitor has beneficial or detrimental effects is dose and time dependent. Thus, the inhibitor bortezomib, indicated for multiple myeloma treatment, may demonstrate cardiovascular toxicity.
The UPS becomes dysfunctional during ischemia, while its function is preserved by ischemic preconditioning.
The UPS exerts a parallel function to AUTO, which also prevents proteotoxic stress, but targets mostly macromolecular structures and organelles. Moreover, AUTO is upregulated while the UPS becomes dysfunctional at ischemia. Actually, it is suggested that there is an inverse relationship between UPS and autophagic flux.
15.4.2 Efforts at CD Modality Clarification
It must be pointed out that the efforts to further clarify the various modalities of CD continue.
Thus the nomenclature Committee on Cell Death has issued Recommendations in 2005 and 2009 [2]. They state that it is important to discriminate between dying as a process and death as an end point (Table 15.1).
They also suggest that exact description of various characteristics should replace the mention of the modality itself i.e. TUNEL positive cells % instead of APO %. Similarly they recommend that “double membraned microvesicles” should be described instead of AUTO.
Galluzzi et al. [1] as many other authors have, describe that N is a negative definition i.e. cell death with no signs of APO or AUTO. Moreover they suggest limiting the use of the term oncosis. However, Kostin [5] extensively uses this term.
The Committee authors [2] also describe the term: Mitotic catastrophe which occurs after a dysregulated of failed mitosis and is morphologically accompanied by micro- and multinucleosis.
Also in their Table 15.2 they describe “cornification”.
Table 15.2
Distinct modalities of cell death
Cell death mode | Morphological features |
|---|---|
Apoptosis | Rounding-up of the cell |
Retraction of pseudopodes | |
Reduction of cellular and nuclear volume (pyknosis) | |
Nuclear fragmentation (karyorrhexis) | |
Minor modification of cytoplasmic organelles | |
Plasma membrane blebbing | |
Engulfment by resident phagocytes, in vivo | |
Autophagy | Lack of chromatin condensation |
Massive vacuolization of the cytoplasm | |
Accumulation of (double-membraned) autophagic vacuoles | |
Little or no uptake by phagocytic cells, in vivo | |
Cornification | Elimination of cytosolic organelles |
Modifications of plasma membrane | |
Accumulation of lipids in F and L granules | |
Extrusion of lipids in the extracellular space | |
Desquamation (loss of corneocytes) by protease activation | |
Necrosis | Cytoplasmic swelling (oncosis) |
Rupture of plasma membrane | |
Swelling of cytoplasmic organelles | |
Moderate chromatin condensation |
They also comment on the validity of the term “programmed cell death”.
They point out that “genetic programming” characterizes APO during development and aging. They recommend that the terms developmental, etoposide, osmotic shock-induced cell death etc. should be used.
Moreover, they give a useful suggestion as to when a cell can be considered dead (Table 15.1).
1.
When it has lost the integrity of its plasma membrane.
2.
When including the nucleolus. it has undergone complete fragmentation into discrete bodies.
3.
Its corpse or its fragments has been engulfed by an adjacent cell in vivo.
In their Table 15.2 they give morphologic characteristics of APO, AUTO, and N.
15.4.3 Cross Talk Among Processes
Before finishing this description of the various forms of CD, the common pathways of the crosstalk among the three processes, which has already been referred to, and addressed by most expert groups [1–3, 12] must be further discussed.
Kung et al. [35] described experiments both in C. elegans and mammalian cells.
They stress that the process of N is as old as APO, and that Virchow first described the “unregulated” or ‘accidental” form of N. They describe that in APO the main mechanism is permeabilization of the MOM, while in N the opening of the MPTP, which spans the MIM. They also point out that the same death ligands that can activate APO (TNFα, Fas ligand TRAIL-TNF-related APO-inducing ligand) can also stimulate N, and that TNFα can induce survival, APO or N. They also remind how, when caspase-8 is deleted or inhibited, APO cannot be initiated and programmed N emerges through TNFR1 ligation. They also remind that apart from Ca2+ overload, Fe2+ cystolic increase can lead to TNFα-induced N. They also remind that APO and N are connected at the death receptor and MITO pathways, and that the MOM and MIM are separated by only microns, promoting these cross-talks.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


