The Kidney in Heart Failure
JoAnn Lindenfeld
Robert W. Schrier
Sodium and water retention are hallmarks of heart failure (HF) and link the heart and kidney in this common medical problem. Only recently has it been recognized that renal dysfunction is an important predictor of morbidity and mortality due to HF. When renal dysfunction occurs in the setting of HF, there is a higher mortality, a more prolonged hospital stay, and a higher rate of readmission for HF (1,2,3,4,5). However, it remains uncertain whether renal dysfunction occurring with HF is merely a reflection of the severity of the HF or whether the renal dysfunction actually accelerates the progression of HF (6). In this chapter we review what is known about renal salt and water handling in HF and describe the clinical characteristics of the so-called cardiorenal syndrome. We then explore potential mechanisms whereby renal dysfunction may exacerbate the progression of HF and address potential therapeutic strategies to ameliorate worsening renal function in patients with HF.
Renal Salt and Water Handling in Heart Failure
A substantial body of evidence supports a unifying hypothesis of body fluid volume regulation in health and disease (7,8,9). While the exact nature of this volume-regulatory system remains incompletely understood, the factors involved in renal salt and water handling in HF can be broadly classified as belonging to either afferent (sensor) or efferent (effector) limbs. This integrated mechanism provides an understanding of why patients with HF have expanded total plasma and blood volumes and yet their kidneys, which are otherwise normal, continue to retain sodium and water.
Afferent Mechanisms
The Concept of Adequate Arterial Filling or the Compartment Sensed
There are a number of circumstances in which there is avid renal sodium and water retention despite expanded extracellular fluid (ECF), interstitial fluid (ISF), and intravascu-lar volumes. For example, patients with advanced HF often have increased ECF volumes, including interstitial edema, expanded total plasma, and blood volumes (7,8,9). In fact, the edematous disorders, including HF, cirrhosis, and pregnancy are defined by avid renal sodium and water retention despite total body salt and water excess. In these disease states, it is clear that the integrity of the kidney as the ultimate effector organ of body fluid regulation is intact. For example, renal sodium and water retention in cirrhotic patients is reversed by transplantation of the kidney from a cirrhotic patient to a subject with normal liver function (10). Moreover, the transplantation of a normal liver into the edematous cirrhotic patient or implantation of a left ventricular assist device into an edematous HF patient have both been shown to reverse renal sodium and water retention (11,12). Thus, in such edematous disorders the kidney must be responding to extrarenal signals from the afferent limb of a volume-regulatory system. The pathogenesis of these extrarenal signals, although not completely understood, is discussed next.
If afferent volume receptors sense primarily total blood volume, then the kidneys of edematous patients should increase their excretion of sodium and water as total blood volume increases. Since this does not occur in patients with severe cardiac or liver disease, there must be some body fluid compartment that remains underfilled even in the presence of expansion of total ECF and blood volume. This underfilled compartment is the afferent limb of renal sodium and water retention in patients with ede-matous disorders. In 1948, John Peters at Yale coined the term effective blood volume as a reference to just such an underfilled body fluid compartment (13). Peters postulated that a decrease in effective blood volume triggers extrarenal signals that enhance tubular sodium and water reabsorption by the otherwise normal kidney. Indeed, renal sodium and water retention can occur in patients with cardiac failure prior to any diminution in glomerular filtration rate (GFR).
Borst and De Vries (14) first suggested that cardiac output was the effective blood volume postulated by Peters and, as such, was the primary modulator of renal sodium and water excretion. However, cardiac output is unlikely the only candidate for effective blood volume because profound renal salt and water retention may occur in the presence of increased in cardiac output. Cirrhosis, pregnancy, large arteriovenous (AV) fistulas, and other causes of high-output cardiac failure such as thyrotoxicosis and beriberi are all associated with significant elevations in cardiac output and avid renal sodium and water retention with expansion of ECF volume (7,8,9).
Primacy of the Arterial Circulation in Volume Regulation
A unifying hypothesis for body fluid volume regulation in health and disease (7,8,9) has been developed from a series of investigations in experimental animals and in humans (15,16,17,18,19,20,21,22,23,24,25). This unifying hypothesis states that total ECF, ISF, or intravascular volumes are not primary determinants of renal sodium and water excretion. With this hypothesis, the venous component of intravascular volume is likewise excluded as the primary determinant of sodium and water excretion. However, it is acknowledged that there are experimental and clinical circumstances in which selective increases in right and left or only left atrial pressure stimulate the release of atrial natriuretic peptide (ANP) (9) and suppression of arginine vasopressin (AVP) (26), respectively, which may enhance sodium and water excretion. However, these events must be subservient to more potent determinants of body fluid volume regulation because the patient with advanced left or right ventricular dysfunction, or both, exhibits avid sodium and water retention despite markedly elevated atrial pressures.
The unifying hypothesis of body fluid volume regulation proposes that the arterial circulation is the primary body fluid compartment modulating renal sodium and water excretion (7,8,9). In a 70-kg man, total body water approximates 42 L, of which only 0.7 L (1.7% of total body water) resides in the arterial circulation (Table 15-1). From a teleologic viewpoint, it is attractive to propose that the primary regulation of renal sodium and water excretion, and thus body fluid volume homeostasis, is modulated by the smallest body fluid compartment, thus endowing the system with exquisite sensitivity to relatively small changes in body fluid volume, and resides in that fluid compartment responsible for the arterial perfusion of the body’s vital organs and tissues.
Table 15-1 Body Fluid Volume Distribution | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Cardiac Output and Peripheral Arterial Resistance as Determinants of Arterial Filling
The body fluid volume regulation hypothesis proposes that cardiac output and peripheral vascular resistance are the two primary determinants of overfilling or underfilling of the arterial circulation. In this context, it is proposed that all renal sodium-and water-retaining states, which occur in the absence of intrinsic renal disease, are initiated by either a decrease in cardiac output, peripheral arterial vasodilation, or both. Thus, this hypothesis accounts for sodium and water retention in both low-output and high-output cardiac failure as well as in other edematous disorders with intact kidney function.
Afferent Volume Receptors
The afferent volume receptors for such a volume regulatory system must reside in the arterial vascular tree, such as the high-pressure baroreceptors in the carotid sinus, aortic arch, left ventricle, and juxtaglomerular apparatus. As previously mentioned, the low-pressure volume receptors of the thorax (atria, right ventricle, and pulmonary vessels) must be of some importance to the volume regulatory system. However, there is considerable evidence that arterial receptors predominate over low-pressure receptors in volume control in mammals (27,28,29,30,31,32,33,34,35).
Low-pressure Volume Receptors
Various maneuvers that decrease central venous return, such as positive pressure breathing (36), lower extremity tourniquets (37,38), and prolonged standing (39) are associated with decreased renal sodium excretion. Conversely, maneuvers that increase thoracic venous return, such as negative pressure breathing (40) and recumbency (41), are
associated with enhanced renal sodium excretion. Headout water immersion, a technique that increases venous return to the heart, results in a significant increase in renal salt and water excretion independent of major changes in either GFR or renal hemodynamics (42). Moreover, in the dog a direct correlation between renal sodium excretion and left atrial pressure has been demonstrated (43,44), supporting a role for an atrial receptor in volume regulation. As first suggested by Gauer and Henry (40,45), physiologically important left atrial receptors have also been shown to contribute to ECF volume regulation by exerting nonosmotic control over the antidiuretic hormone AVP (26, 43, 45). In addition, changes in atrial stretch and transmural pressures have been shown to determine circulating plasma concentrations of the vasoactive and natriuretic hormone ANP (46,47,48). Another normal response to increased left atrial pressure is a decrease in renal sympathetic tone (49). In chronic HF these atrial-renal reflexes are impaired. For example, the atrial-renal sympathetic reflex is also blunted in a dog model of HF (50). In patients with dilated cardiomyopathy and mild HF, a volume load failed to increase ANP and the natriuretic response was blunted (51).
associated with enhanced renal sodium excretion. Headout water immersion, a technique that increases venous return to the heart, results in a significant increase in renal salt and water excretion independent of major changes in either GFR or renal hemodynamics (42). Moreover, in the dog a direct correlation between renal sodium excretion and left atrial pressure has been demonstrated (43,44), supporting a role for an atrial receptor in volume regulation. As first suggested by Gauer and Henry (40,45), physiologically important left atrial receptors have also been shown to contribute to ECF volume regulation by exerting nonosmotic control over the antidiuretic hormone AVP (26, 43, 45). In addition, changes in atrial stretch and transmural pressures have been shown to determine circulating plasma concentrations of the vasoactive and natriuretic hormone ANP (46,47,48). Another normal response to increased left atrial pressure is a decrease in renal sympathetic tone (49). In chronic HF these atrial-renal reflexes are impaired. For example, the atrial-renal sympathetic reflex is also blunted in a dog model of HF (50). In patients with dilated cardiomyopathy and mild HF, a volume load failed to increase ANP and the natriuretic response was blunted (51).
High-Pressure Volume Receptors
Evidence for the presence of volume-sensitive receptors in the arterial circulation in humans originated from observations in subjects with traumatic AV fistulas (52). Closure of an AV fistula is associated with a decreased rate of emptying of the arterial blood into the venous circulation, as demonstrated by closure-induced increases in diastolic arterial pressure and decreases in cardiac output, and results in an immediate increase in renal sodium excretion without changes in either GFR or renal blood flow (RBF) (52). Further evidence implicating the fullness of the arterial vascular tree as a sensor in modulating renal sodium excretion can be found in denervation experiments. In these studies, pharmacologic or surgical interruption of sympathetic afferent neural pathways emanating from high-pressure areas inhibited the natriuretic response to volume expansion (15,16,53,54,55,56,57). Moreover, reduction of pressure or stretch at the carotid sinus has been shown to activate the sympathetic nervous system and to promote renal sodium and water retention (58,59). High-pressure baroreceptors also appear to be important factors in regulating the nonosmotic release of AVP and, thus, renal water excretion (60,61).
Located in the afferent arterioles within the kidney, the juxtaglomerular apparatus is one of the best-defined of the high-pressure receptors implicated in body fluid volume regulation. The juxtaglomerular apparatus responds to decreased stretch or increased renal sympathetic tone with enhanced renal secretion of renin (59). Thus, this baroreceptor is an important factor in the control of angiotensin II formation and aldosterone secretion and, ultimately, in renal sodium and water retention.
Other Afferent Receptors
While unloading of high-pressure volume receptors appears to play the predominant role in initiating renal salt and water retention in HF and in other edematous disorders with intact kidney function, experimental evidence suggests a possible role for various chemoreceptors found in the heart and elsewhere in the body. For example, in the heart and lungs, both vagal and sympathetic afferent nerve endings respond to a variety of exogenous and endogenous chemical substances, including capsaicin, phenyldiguanidine, bradykinin, substance P, and prostaglandins (62,63,64). Significantly, Zucker et al. (63) have shown that prostaglandin I2 (PGI2) attenuates the baroreflex control of renal nerve activity via a cardiac afferent vagal mechanism. Because substances such as bradykinin and prostaglandins may circulate at increased concentrations in HF patients (65), it is possible that altered central nervous system input from chemically sensitive cardiac or pulmonary afferents contributes to the sodium and water retention of chronic HF. However, the exact role in HF of these and other chemoreceptors remains to be defined.
Efferent Mechanisms
Renal Hemodynamics
The GFR usually is normal in early or mild HF and is reduced only as cardiac performance becomes more severely impaired. Renal vascular resistance is increased with a concomitant decrease in RBF. In general, RBF decreases in proportion to the decrease in cardiac performance. Thus, the ratio of GFR to renal plasma flow, or the filtration fraction, usually is increased in patients with cardiac failure. This increased filtration fraction is a consequence of constriction of the efferent arterioles within the kidney. These changes in renal hemodynamics alter the hydrostatic and oncotic forces in the peritubular capillaries to favor increased proximal tubular reabsorption of sodium and water (Fig. 15-1). The renal hemodynamic changes seen in HF are primarily mediated by activation of various neurohormonal vasoconstrictor
systems. In addition, neurohormonal activation directly contributes to enhanced sodium and water reabsorption in both the proximal and distal nephron in HF, as discussed next.
systems. In addition, neurohormonal activation directly contributes to enhanced sodium and water reabsorption in both the proximal and distal nephron in HF, as discussed next.
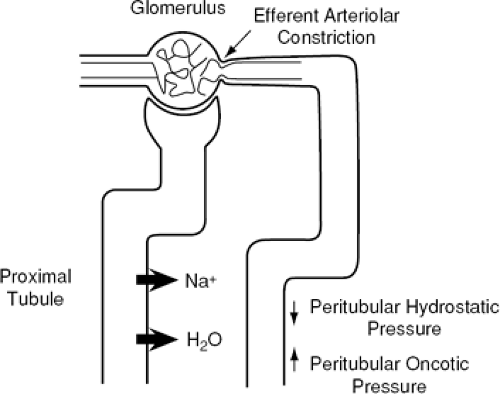 Figure 15-1 Efferent arteriolar constriction, mediated in congestive heart failure (CHF) by increased renal sympathetic nerve activity and angiotensin II concentrations, alters net postglomerular Starling forces in a direction to enhance proximal tubular sodium and water reabsorption. |
The Neurohormonal Response to Heart Failure
Arterial underfilling secondary to a diminished cardiac output elicits a number of compensatory neurohormonal responses that act to maintain the integrity of the arterial circulation by promoting peripheral vasoconstriction as well as expansion of the ECF volume through renal sodium and water retention. The three best-described neurohor-monal vasoconstrictor systems activated in response to cardiac failure are the sympathetic nervous system, the reninangiotensin-aldosterone system (RAAS), and the nonosmotic release of AVP. Baroreceptor activation of the sympathetic nervous system appears to be the primary integrator of the hormonal vasoconstrictor systems involved in the volume control system because the nonosmotic release of AVP involves sympathetic stimulation of the supraoptic and paraventricular nuclei in the hypothalamus (66), and activation of the RAAS involves renal adrenergic stimulation (67). Thus, in low-output cardiac failure, diminished integrity of the arterial circulation, as determined by decreased cardiac output, causes unloading of arterial baroreccptors in the carotid sinus and aortic arch. Peripheral vasodilation causes unloading of these arterial baroreceptors in the setting of high-output cardiac failure (Fig. 15-2). This baroreceptor inactivation results in diminution of the tonic inhibitory effect of afferent vagal and glossopharyngeal pathways to the central nervous system and initiates an increase in sympathetic efferent adrenergic tone with subsequent activation of the other two major vasoconstrictor hormonal systems. Various counterregulatory vasodilatory and natriuretic hormones are also activated in HF, including the natriuretic peptides (NPs) and renal prostaglandins. Table 15-2 summarizes the renal effects of some of the neurohormonal systems that are activated in HF.
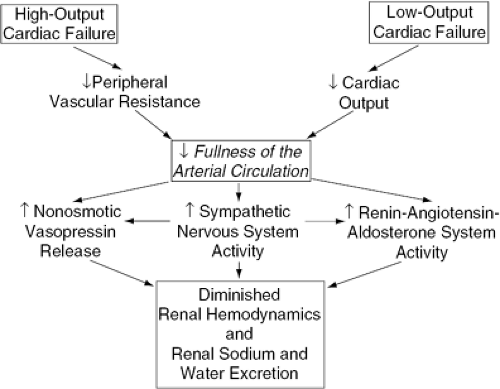 Figure 15-2 Proposed mechanism of renal sodium and water retention in high-output and low-output cardiac failure. |
Table 15-2 Renal Effects of Neurohormonal Activation in Congestive Heart Failure | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The Sympathetic Nervous System in Heart Failure
The sympathetic nervous system is activated early in patients with HF. Evidence for this comes both from indirect and direct measurements of sympathetic nervous system activity in HF patients. Various studies have documented elevated peripheral venous plasma norepinephrine (NE) concentrations in patients with HF (68,69,70,71,72,73). Previous studies in advanced HF, using tritiated NE to determine NE kinetics, demonstrated that both increased NE secretion and decreased NE clearance contribute to the high venous plasma NE concentrations seen in these patients, suggesting that increased sympathetic activity is at least partially responsible for the elevated circulating plasma NE (70,71). A more recent investigation of NE kinetics in earlier stages of HF demonstrated that the initial increase in plasma NE in HF is solely due to increased NE secretion, providing evidence of increased sympathetic nervous activity early in
the course of HF (72). Studies using peroneal nerve microneurography to directly assess sympathetic nerve activity to muscle also demonstrate increased sympathetic activity in HF patients. Using this technique, one group of investigators recorded sympathetic nerve activity to muscle while simultaneously measuring plasma NE concentrations in patients with HF (74). Sympathetic nerve activity was increased in these patients and strongly correlated with plasma NE levels (74). Finally, the SOLVD Investigators (75) have reported the presence of sympathetic activation, as determined by plasma NE concentrations, in patients with asymptomatic LV dysfunction.
the course of HF (72). Studies using peroneal nerve microneurography to directly assess sympathetic nerve activity to muscle also demonstrate increased sympathetic activity in HF patients. Using this technique, one group of investigators recorded sympathetic nerve activity to muscle while simultaneously measuring plasma NE concentrations in patients with HF (74). Sympathetic nerve activity was increased in these patients and strongly correlated with plasma NE levels (74). Finally, the SOLVD Investigators (75) have reported the presence of sympathetic activation, as determined by plasma NE concentrations, in patients with asymptomatic LV dysfunction.
A study in human HF have demonstrated the presence of selective cardiorenal sympathetic activation (70). In this study of whole-body and organ-specific NE kinetics in HF patients, cardiac and renal NE spillover rates were increased 504% and 206%, respectively, whereas the NE spillover rate from the lungs was normal. Thus, it is perhaps not surprising that the deleterious effects of chronic sympathetic activation in HF are seen primarily in the heart and kidney. The importance of increased renal sympathetic nerve activity was demonstrated in a recent study of 61 HF patients. In a model that included total body NE spillover, left ventricular ejection fraction (LVEF), GFR, RBF, cardiac index, etiology of HF, and age, renal NE spillover was the most important predictor of all-cause mortality and need for heart transplantation.
The adverse effects of systemic and cardiac adrenergic activation in chronic HF include peripheral vasoconstriction, renal sodium and water retention, direct myocardial toxicity via calcium overload or the induction of apoptosis, stimulation of tachycardia and other arrhythmias, activation of the RAAS, stimulation of nonosmotic AVP release, and downregulation of cardiac β1-adrenergic receptors and other myocardial receptor-signal transduction abnormalities. This latter effect, which occurs in response to increased cardiac sympathetic activity, may be alternatively viewed as a protective mechanism to withdraw the heart from the effects of excessive adrenergic stimulation. Sympathetic activation, as measured by the plasma NE concentration, correlates directly with the degree of LV dysfunction in HF (68,69,76). Moreover, the plasma NE concentration correlates negatively with prognosis in HF (77). That is, high plasma NE levels are associated with poor prognosis in HF patients. This was demonstrated by Cohn et al. (77), who prospectively studied 106 patients with moderate or severe HF and found that a single resting venous plasma NE level provided a better guide to prognosis than did other commonly measured indexes of cardiac and functional performance. Recent studies confirm the predictive value of plasma NE even in patients treated with angiotensin-converting enzyme (ACE) inhibitors and β-blockers (78). Finally, β-adrenergic receptor antagonists have been demonstrated to improve LVEF and mortality and reduce hospital admissions in patients with HF (79,80,81). Taken together, these observations support the hypothesis that NE contributes to disease progression in chronic HF.
Effects of Increased Renal Sympathetic Activity in Heart Failure
Through renal vasoconstriction, stimulation of the RAAS, and direct effects on the proximal convoluted tubule, enhanced renal sympathetic activity may contribute to the avid sodium and water retention of HF. Indeed, intrarenal adrenergic blockade has been shown to cause a natriuresis in experimental animals and in humans with HF (82,83,84). Moreover, in the rat, renal nerve stimulation has been demonstrated to produce an approximate 25% reduction in sodium excretion and urine volume (85). The diminished renal sodium excretion that accompanies renal nerve stimulation may be mediated by at least two mechanisms. Studies performed in rats have demonstrated that NE-induced efferent arteriolar constriction alters peritubular hemodynamic forces in favor of increased tubular sodium reabsorption (86). As previously mentioned, the increase in filtration fraction with a normal or only slightly reduced GFR that is often seen in HF patients must be due to predominant efferent arteriolar constriction. Constriction of the efferent arterioles in HF has been confirmed by renal micropuncture studies performed in rats (87), and is at least partially mediated by increased renal sympathetic activity and by angiotensin II. Thus, efferent arteriolar constriction in HF alters the balance of hemodynamic forces in the peritubular capillaries in favor of enhanced proximal tubular sodium reabsorption.
In addition, renal nerves have been shown to exert a direct influence on sodium reabsorption in the proximal convoluted tubule (85,88). Bello-Reuss et al. (85) demonstrated this direct effect of renal nerve activation to enhance proximal tubular sodium reabsorption in wholekidney and individual nephron studies in the rat. In these animals, renal nerve stimulation produced an increase in the tubular fluid/plasma inulin concentration ratio in the late proximal tubule, an outcome of increased fractional sodium and water reabsorption in this segment of the nephron (85). Hence, increased renal nerve activity may promote sodium retention by a mechanism independent of changes in renal hemodynamics. However, in dogs with denervated transplanted kidneys and chronic vena caval constriction, the sodium retention persists (89). Moreover, renal denervation does not prevent ascites in the dog with chronic vena caval constriction (90). Thus, renal nerves probably contribute to, but do not fully account for, the avid sodium and water retention of HF.
The Renin-Angiotensin-Aldosterone System in Heart Failure
The RAAS is also activated in HF, as assessed by plasma renin activity (PRA) (69,91,92). Renin acts on angiotensinogen to produce angiotensin I, which is then converted by ACE to angiotensin II. In HF, the resultant increased plasma concentration of angiotensin II exerts circulatory effects similar to sympathetic activation, including peripheral arterial and venous vascular constriction, renal vasoconstriction, and cardiac inotropism. Angiotensin II also acts to promote the secretion of the sodium-retaining hormone aldosterone by the adrenal cortex and in positive-feedback stimulation of the sympathetic nervous system (93). Thus, in the kidney, activation of this hormonal system promotes sodium retention via several mechanisms, as discussed later.
Activation of the RAAS is associated with hyponatremia and an unfavorable prognosis in HF (65,94). The association
of PRA and hyponatremia was first described by Dzau et al. (65) in a cohort of 15 HF patients. These data demonstrated that normal or suppressed PRA is associated with a normal serum sodium level, whereas the highest PRA is associated with the lowest serum sodium concentrations (65). This association of hyponatremia with RAAS activation was later confirmed by Lee and Packer (94) in a larger group of HF patients. Moreover, these investigators demonstrated the association of this hyponatremic, hyperreninemic state with poor survival (94). The mechanisms by which activation of the RAAS might negatively impact on survival in HF are primarily mediated by increased angiotensin II levels. As previously noted, increased angiotensin II causes venous and arteriolar vasoconstriction, thus increasing ventricular preload and afterload. Angiotensin II may stimulate nonosmotic AVP release, promoting renal water retention, and increase thirst. Angiotensin II also may act as a growth factor, promoting myocardial hypertrophy and thus ventricular ischemia and remodeling, as well as vascular and glomerular hypertrophy and remodeling. Angiotensin II, via several mechanisms, augments renal sodium reabsorption.
of PRA and hyponatremia was first described by Dzau et al. (65) in a cohort of 15 HF patients. These data demonstrated that normal or suppressed PRA is associated with a normal serum sodium level, whereas the highest PRA is associated with the lowest serum sodium concentrations (65). This association of hyponatremia with RAAS activation was later confirmed by Lee and Packer (94) in a larger group of HF patients. Moreover, these investigators demonstrated the association of this hyponatremic, hyperreninemic state with poor survival (94). The mechanisms by which activation of the RAAS might negatively impact on survival in HF are primarily mediated by increased angiotensin II levels. As previously noted, increased angiotensin II causes venous and arteriolar vasoconstriction, thus increasing ventricular preload and afterload. Angiotensin II may stimulate nonosmotic AVP release, promoting renal water retention, and increase thirst. Angiotensin II also may act as a growth factor, promoting myocardial hypertrophy and thus ventricular ischemia and remodeling, as well as vascular and glomerular hypertrophy and remodeling. Angiotensin II, via several mechanisms, augments renal sodium reabsorption.
Renal Effects of Increased Angiotensin II in Heart Failure
Angiotensin II may contribute to the sodium and water retention of HF through direct and indirect effects on proximal tubular sodium reabsorption and by stimulating the release of aldosterone from the adrenal gland. Angiotensin II causes renal efferent arteriolar vasoconstriction, resulting in decreased RBF and an increased filtration fraction. As with renal nerve stimulation, this results in increased peritubular capillary oncotic pressure and reduced peritubular capillary hydrostatic pressure, which favors the reabsorption of sodium and water in the proximal tubule (87,95). In addition, angiotensin II has been shown to have a direct effect on enhanced sodium reabsorption in the proximal tubule (96,97). This direct effect of angiotensin II on the proximal tubule has been associated with angiotensin II-mediated activation of the basolateral sodium-bicarbonate cotransporters and apical sodium-hydrogen exchanger and is inhibited by angiotensin II AT1-receptor blockade (97). Finally, angiotensin II enhances aldosterone secretion by the adrenal gland, which promotes tubular sodium reabsorption in the distal tubule and collecting duct.
A role for RAAS activation in the sodium retention of HF is suggested by the finding that urinary sodium excretion inversely correlates with PRA and urinary aldosterone excretion in HF patients (98,99). However, the administration of an ACE inhibitor during HF does not consistently increase urinary sodium excretion, despite a consistent decrease in plasma aldosterone concentration (100). The simultaneous decrease in blood pressure due to decreased circulating concentrations of angiotensin II, however, may activate hemodynamic and neurohormonal mechanisms, which could obscure the natriuretic response to lowered angiotensin II and aldosterone concentrations.
Support for this hypothesis comes from a study performed by Hensen et al. (101). The effect of the specific aldosterone antagonist, spironolactone, on urinary sodium excretion was examined in patients with mild to moderate HF who were withdrawn from all medications prior to study. Sodium was retained in all patients throughout the period prior to aldosterone antagonism. On an average sodium intake was 97 ± 8 mmol per day, and the average sodium excretion prior to spironolactone was 78 ± 8 mmol per day. During therapy with spironolactone, all HF patients exhibited a significant increase in urinary sodium excretion to an average of 131 ± 13 mmol per day. Plasma renin activity and NE increased during the administration of spironolactone. Thus, this investigation demonstrates reversal of the sodium retention of HF with the administration of an aldosterone antagonist, despite further activation of various antinatriuretic influences, including stimulation of the renin-angiotensin and sympathetic nervous systems. These results indicate that the RAAS is an important mediator of sodium retention in HF.
The Nonosmotic Release of Vasopressin in Heart Failure
Plasma AVP is usually elevated in patients with HF and correlates in general with the clinical and hemodynamic severity of disease and with the serum sodium level (18,102,103,104,105). Plasma AVP concentrations are elevated in spite of hypo-osmality in patients with HF, and these levels fail to suppress normally to acute water loading (102,106). Taken together, these observations suggest that there is enhanced release of AVP in HF and that nonosmotic mechanisms are responsible. As already suggested, baroreceptor activation of the sympathetic nervous system likely mediates the nonosmotic release of AVP in HF patients (66).
Through its effects on vascular (V1) and renal (V2) receptors, AVP is a potent mediator of peripheral vasoconstriction and renal water retention, respectively. By analogy to NE and angiotensin II, it can be speculated that both of these effects may be of some pathophysiological importance in HF. Several findings support a role for AVP in the altered hemodynamics of HF. For example, small increases in plasma AVP concentrations, within the basal range seen in HF patients, increase peripheral vascular resistance and produce a corresponding decrease in cardiac output in patients with cardiac failure (105). Moreover, experimental antagonists of the V1 vasopressin receptor have been used in patients with lowoutput cardiac failure (107,108). Selective antagonism of these receptors in humans is associated with peripheral vasodilation and improved cardiac function in a subset of patients with severe HF (107,108). Finally, it has been shown that a nonpeptide V1 receptor antagonist decreases systemic vascular resistance and increases cardiac output in dogs with tachycardia-induced HF (109). These observations suggest a role for AVP in the peripheral vasoconstriction and, hence, increased afterload of HF.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


