Fig. 6.1
The HPA axis and systemic actions of glucocorticoids. CRH corticotropin-releasing hormone, ACTH adrenocorticotropic hormone (Figure 6.1 was prepared using image vectors from Servier Medical Art (www.ervier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/))
6.1.3 Glucocorticoids and Glucocorticoid Receptor
Glucocorticoids, the end-products of the HPA axis, play a fundamental role in the maintenance of basal and stress-related homeostasis, and regulate a broad spectrum of physiologic functions, such as the cardiovascular tone, the intermediary metabolism primarily through catabolic actions, the quantity and the quality of the inflammatory and immune response and many other functions, by altering the transcription rate of a significant percentage of genes in a positive or negative way [3, 4, 8] (Fig. 6.1). Glucocorticoids exert their diverse actions through an intracellular, ubiquitously expressed protein, the human glucocorticoid receptor (hGR), which belongs to the nuclear receptor superfamily of transcription factors [16].
The hGR gene is located on the short arm of chromosome 5 and consists of 10 exons [16]. The alternative use of two distinct exons, 9α and 9β, generates two main isoforms, the hGRα and the hGRβ [17]. The hGRα is a 777-amino acid protein and represents the classic isoform, which binds endogenous and synthetic glucocorticoids, and mediates all genomic effects of these hormones in almost every cell type in the human organism [17]. On the other hand, the hGRβ contains 742 amino acids, does not bind glucocorticoids, and acts as a dominant negative regulator on the hGRα-mediated transcriptional activity through several mechanisms, such as heterodimerization and competition with hGRα for binding to glucocorticoid response elements (GREs) in the promoter regions of target genes, or for interacting with nuclear receptor coregulators (coactivators, corepressors) [17–21]. Furthermore, the hGRα mRNA was demonstrated to be translated from eight alternative initiation sites into functionally distinct hGRα protein isoforms by ribosomal leaky scanning or ribosomal shunting [22]. Given that hGRβ mRNA translation is initiated by the same 5′ termini, it is possible that the above translation mechanisms could give rise to eight different hGRβ proteins as well [8]. Thus, the 16 amino terminal hGRα and hGRβ protein isoforms may form 256 homo- or hetero-dimers to transduce the glucocorticoid signal.
At the cellular level, hGRα is primarily localized in the cytoplasm of the target cell in the absence of a ligand, as part of a multiprotein complex consisting of heat shock proteins and immunophilins [16] (Fig. 6.2). Upon ligand-binding, hGRα undergoes conformational change, dissociates from the rest of proteins, and translocates into the nucleus through the nuclear pore, via an active ATP-dependent process mediated by its nuclear localization signal (NL)-1 and −2 [19]. Within the nucleus, the ligand-activated GR directly interacts as a homo- or hetero-dimer with specific DNA sequences, the glucocorticoid response elements (GREs), in the promoter regions of target genes [19]. The GR contains two transactivation domains, activation function (AF)-1 and −2, located at its NTD and LBD, respectively, through which it interacts with protein complexes, such as the nuclear receptor coactivators p160, p300/CREB-binding protein (CBP) and p300/CBP-associated factor (p/CAF) complexes and the SWI/SNF and vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein (DRIP/TRAP) chromatin-remodeling complexes; it influences the activity of the RNA polymerase II and its ancillary factors, thereby modulating the transcription rates of glucocorticoid-responsive genes [16] (Fig. 6.2).
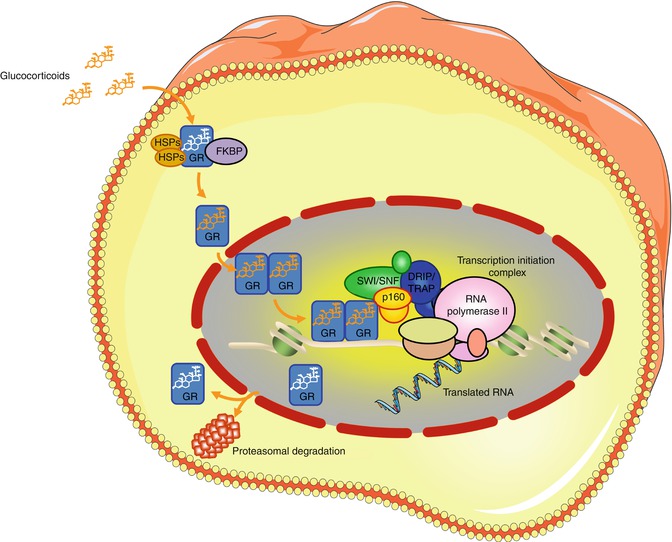
Fig. 6.2
Glucocorticoid signaling pathway. GR glucocorticoid receptor, HSP heat shock proteins, FKBP immunophilins, p160 nuclear receptor coactivators p160, SWI/SNF switching/sucrose non-fermenting complex, DRIP/TRAP vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein complex (Figure 6.2 was prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/))
Alternatively, hGRα may modulate gene expression independently of its DNA binding, by interacting with and altering the functions of other known transcription factors, such as NF-κB, STAT-5, and AP-1, possibly as monomer [16]. These interacting factors, in turn, may alter the actions of the glucocorticoid receptor acting on positive or negative GREs.
6.1.4 Disorders of the HPA Axis
The disorders of the HPA axis can be categorized according to the anatomical component(s) that is/are primarily affected: (1) the hypothalamus, (2) the pituitary gland, (3) the adrenal glands and (4) other tissues and organs, including alterations of glucocorticoid availability or action at target tissues (Fig. 6.3). These disorders may result in either elevated or reduced concentrations or effects of circulating glucocorticoids; thus, symptoms and signs are associated with increased or decreased actions of these hormones in the responsive tissues, (Table 6.1).
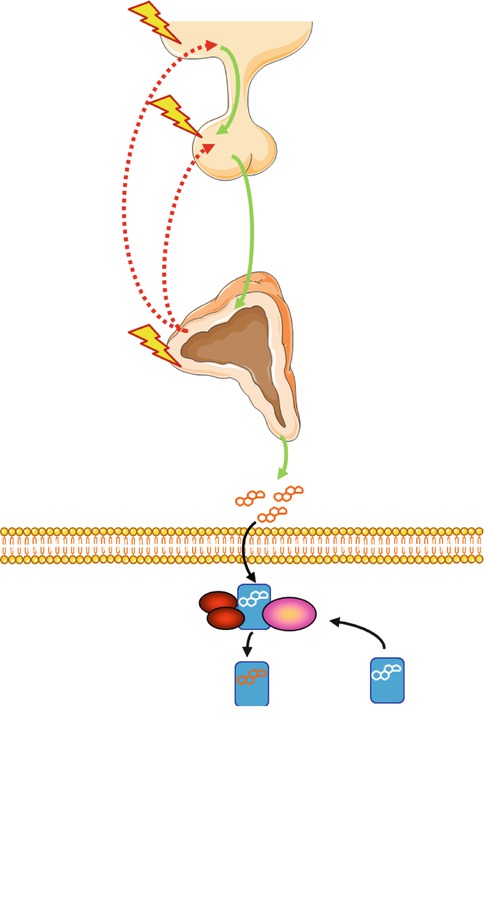
Fig. 6.3
The HPA axis and associated disorders. The disorders of the HPA axis can be categorized according to the anatomical component(s) that is/are primarily affected: (1) the hypothalamus, (2) the pituitary gland, (3) the adrenal glands and (4) the glucocorticoid-responsive cell. Mutations or polymorphisms of the hGR gene may impair normal glucocorticoid signal transduction, thus altering tissue sensitivity to glucocorticoids. CRH corticotropin-releasing hormone, ACTH adrenocorticotropic hormone, GR glucocorticoid receptor (Figure was prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/))
Table 6.1
Expected clinical manifestations in tissue-specific glucocorticoid excess or hypersensitivity and deficiency or resistance
Target tissue | Glucocorticoid hypersensitivity= Glucocorticoid excess | Glucocorticoid resistance= Glucocorticoid deficiency |
|---|---|---|
Central nervous system | Insomnia, anxiety, depression, defective cognition | Fatigue, somnolence, malaise,defective cognition |
Liver | + Gluconeogenesis, + lipogenesis | Hypoglycemia, resistance to diabetes mellitus |
Fat | Accumulation of visceral fat (metabolic syndrome) | Loss of weight, resistance to weight gain |
Blood vessels | Hypertension | Hypotension |
Bone | Stunted growth, osteoporosis | |
Inflammation/immunity | Immune suppression, anti-inflammation,vulnerability to certain infections and tumors | + Inflammation,+ autoimmunity,+ allergy |
Indeed, chronic glucocorticoid excess results in central obesity, buffalo hump, purple striae of the skin, hypertension, osteoporosis, glucose intolerance/overt diabetes mellitus, dyslipidemia and leukocytosis, all of which are referred to as the “Cushing phenotype” [23]. Patients with pathologically increased concentrations of cortisol are at great risk of severe cardiovascular complications, such as ischemic heart/cerebral disease [23]. Moreover, they are more susceptible to infections secondary to any pathogens, since cortisol suppresses strongly the activity of the immune system.
On the other hand, patients with isolated glucocorticoid deficiency or adrenal insufficiency including aldosterone deficiency may present with fatigue, vomiting, nausea, hypotension, hypoglycemia, hyponatremia, hyperkalemia in the absence of aldosterone secretion and eosinophilia [23]. The decreased concentrations of cortisol lead to compensatory elevations in plasma ACTH concentrations, which result in hyperpigmentation of the skin and buccal mucosa in these patients.
6.1.4.1 Hypothalamic Disorders Associated with Alterations in the HPA Axis
Many space-occupying lesions of the hypothalamus, including craniopharyngioma, Rathke’s pouch cysts, head trauma, inflammatory or infiltrative diseases (encephalitis, meningitis, sarcoidosis, histiocytosis X) and brain malignant tumors (primary or metastatic) lead to destruction of the PVN neurons producing CRH or to dissection of the pituitary stalk that transfers CRH from the hypothalamus to the pituitary gland, thus causing decreased function of the HPA axis [24]. These conditions are often associated with dysfunction of other endocrine axes, such as the hypothalamic-pituitary-gonadal or thyroid axes and the growth hormone/insulin-like growth factor-I axis.
Prader-Willi syndrome is a rare genetic disorder, which is characterized by symptoms associated with hypothalamic dysfunction, such as hyperphagia, obesity, sleep disorders, hypogonadism, hypocortisolism, learning disabilities/borderline intellectual functioning and hypotonia [25].
Patients with the melancholic type of major depression show hyperactivity of the HPA axis and have increased cerebrospinal fluid CRH and norepinephrine, plasma ACTH and serum cortisol concentrations [26, 27]. This sustained hyperfunction of the HPA axis leads to central obesity, hypertension, impaired glucose tolerance, decreased libido and osteoporosis, all of which result in impaired quality of life.
One of the major eating disorders, anorexia nervosa, is also characterized by elevated cortisol concentrations via activation of the HPA axis caused by strict and prolonged energy restriction, as well as by mental stress [28]. Patients with anorexia nervosa demonstrate increased concentrations of CRH in their cerebrospinal fluid [28]. However, in contradistinction to depressed patients, they do not show any clinical manifestations suggestive of glucocorticoid excess. Indeed, they present with symptoms and signs associated with reduced action of glucocorticoids in target tissues, possibly due to lack of food substrate and changes of proteins that may influence the actions of glucocorticoids through the hGR, such as the AMP-activated protein kinase (AMPK) [29].
Any conditions associated with uncoupling of the cortisol circadian rhythm that is under the influence of the SCN CLOCK system, and the circadian peripheral tissue sensitivity to glucocorticoids, under the control of the peripheral CLOCK, may cause the development of components of the metabolic syndrome and its detrimental cardiovascular complications [10, 30]. Night-shift workers and people frequently exposed to jet lag are at an increased risk for developing symptoms and signs of chronic hypercortisolemia, such as central obesity, insulin resistance, dyslipidemia and hypertension, which represent the cardinal clinical manifestation of metabolic syndrome. These pathologic conditions were recently explained at the molecular and cellular level, when it was convincingly shown that the transcription factor “circadian locomotor output cycle kaput” (CLOCK) physically interacts with GR, and catalyzes enzymatically the acetylation of a lysine cluster, located in the hinge region of the receptor and leading to decreased sensitivity to glucocorticoids [10, 30, 31]. This effect of CLOCK on GR can lead to “functional” tissue hypersensitivity to glucocorticoids via uncoupling of circadian cortisol concentrations and peripheral tissue responsiveness to these hormones [10, 30, 31].
6.2 Pituitary Disorders Associated with Alterations in the HPA Axis
6.2.1 Cushing Disease (and Syndrome)
Cushing disease is part of Cushing syndrome, which is characterized by elevated concentrations of circulating cortisol and symptoms and signs of glucocorticoid excess. This disease is caused by semi-autonomous neoplasia or hyperplasia of corticotropes that result in over-secretion of ACTH from the pituitary gland. In contrast, Cushing syndrome indicates a disorder that derives from the adrenal glands, caused by either an adrenocortical adenoma or carcinoma, or from an ectopic site secreting CRH or ACTH (~15 % of ACTH-dependent forms of Cushing disease). Ectopic secretion of ACTH from non-pituitary tissues includes small cell carcinoma of the lung and lung carcinoids, pancreatic or adrenomedullary tumors, as well as other neoplasias [32].
The first manifestation of Cushing disease may be the loss of the diurnal rhythm in ACTH and cortisol secretion. Patients with either Cushing disease or syndrome present with “Cushingoid features”, including moon facies, “buffalo hump”, central obesity, purple skin striae, hypertension, insulin resistance/overt diabetes mellitus, mood changes, menstrual irregularity and osteoporosis [33]. The diagnosis should be made after careful medical history and physical examination, and confirmed by endocrinologic evaluation and imaging studies. Cushing disease is mostly caused by pituitary adenomas. They are usually microadenomas with a diameter less than 1 cm, and are sometimes quite difficult to detect even in the MRI scan with contrast material [34]. Bilateral inferior petrosal sinus sampling with CRH stimulation is helpful in some cases to localize pituitary adenomas in the pituitary including the site of the tumor inside the pituitary gland. The therapeutic approach of patients with Cushing disease starts with resection of the ACTH-secreting pituitary adenoma by transsphenoidal surgery [35]. However, 10–15 % of patients with remission may recur in the future. Bilateral adrenalectomy may be performed in difficult cases in which tumors cannot be successfully resected [35]. Radiation may also be applied to the pituitary gland in combination with medical therapy, usually with mitotane or ketoconazol.
6.2.2 Pituitary Disorders That Result in Hypofunction of the HPA Axis
Any space-occupying lesions in the pituitary gland may lead to hypofunction of the HPA axis and secondary adrenal insufficiency [24]. Pituitary adenomas, cysts, craniopharyngiomas, ependymomas, meningiomas and rarely carcinomas may interfere with ACTH secretion. In addition to these lesions, infections or infiltrative processes, including lymphocytic hypophysitis, haemochromatosis, tuberculosis, meningitis, sarcoidosis, actinomycosis, histiocytosis X and Wegener’s granulomatosis can result in low ACTH release [24]. Sheehan’s syndrome is caused by pituitary apoplexy after labor and consequent pan-hypopituitarism, and used to be a major cause of secondary adrenal insufficiency. However, its incidence has been dramatically reduced because of the improved maternal care in developed countries.
Mutations in genes encoding crucial transcription factors and molecules essential for corticotrope survival and function can cause developmental defects and/or dysfunction in these cells leading to hypofunction of the HPA axis. So far, pathologic mutations have been identified in the genes expressing the transcription factors HESX homolog 1 (HESX1), orthodenticle homeobox 2 (OTX2), LIM homeobox 4 (LHX4), SRY (sex-determining region Y)-box 2 and 3 (SOX2 and SOX3), PROP paired-like homeobox 1 (PROP1) and T-box 19 (TBX19) [36]. Patients harboring these mutations may present with short stature, metabolic disorders, craniofacial abnormalities and learning difficulties/mental retardation associated with abnormalities in the CNS. The onset of adrenal insufficiency occurs generally early in life, although cases owing to PROP1 mutations usually manifest in adolescence.
Congenital pro-opiomelanocortin deficiency is another cause of secondary adrenal insufficiency. The molecular basis of this genetic condition has been ascribed to inactivating mutations in the pro-opiomelanocortin (POMC) gene. Patients develop a characteristic syndrome of hypocortisolism, early onset morbid obesity, and alterations in skin and hair pigmentation [37].
Achalasia-Adrenal Insufficiency-Alacrima syndrome or Allgrove Syndrome, also known as Triple-A syndrome, is a rare autosomal recessive disorder characterized by ACTH-resistant adrenal insufficiency, alacrima (absence of tear secretion), achalasia (dilatation of thoracic esophagus), autonomic dysfunction, and neurodegeneration. This syndrome is caused by mutations in the AAAS gene, which encodes the nuclear pore protein ALADIN with as yet unknown functions [38]. Adrenal insufficiency develops gradually within the first 10 years of life but sometimes becomes evident much later. It may present in some cases as an adrenal crisis with hypoglycemia and seizures.
6.3 Adrenal Disorders Associated with Alterations in the HPA Axis
6.3.1 Cushing Syndrome
Cushing syndrome can be caused by both benign and malignant lesions, such as glucocorticoid-producing adenomas, micro-nodular dysplasia, ACTH-independent macro-nodular hyperplasia or adenocarcinoma of the adrenal cortex [23, 33]. Patients with this syndrome appear with “Cushingoid features”, as described above; however, their clinical manifestations depend on the severity and duration of cortisol excess. Cushing syndrome caused by adenocarcinoma is characterized by large tumors of more than 5 cm in diameter, and are usually rapidly progressing, whereas adenomas are usually small (less than 3 cm) and have generally an insidious onset and chronic course [39]. First choice treatment for Cushing syndrome remains the surgical removal of the tumor. Laparoscopic surgery is currently the approach of choice for the treatment of Cushing syndrome caused by adenomas [40]. In case of failure of resection of the entire mass, usually adenocarcinomas, or those with metastatic lesions, mitotane is used for suppressing both tumor growth and production of steroids [41]. This compound inhibits the cholesterol side-chain cleavage (SCC) enzyme (the human p450 CYP, cholesterol desmolase, or 20, 22 desmolase) and the 11β-hydroxylase (p450 11β or CYP11B1) [41]. Adrenal enzyme inhibitors, such as ketoconazole, metyrapone and etomidate, are also used in clinical practice for patients with surgical failure who remain hypercortisolemic [32].
6.3.2 Adrenal Disorders That Result in Hypofunction of the HPA Axis
Adrenal insufficiency is a disorder first described by Thomas Addison in 1855, which is characterized by deficient production or action of glucocorticoids and/or mineralocorticoids and adrenal androgens [24]. Any pathologic condition associated with destruction of the adrenal cortex can cause primary adrenal insufficiency. The etiology of primary adrenal insufficiency has changed over time. Prior to 1920, the most common cause of primary adrenal insufficiency was tuberculosis, while since 1950, the majority of cases (80–90 %) have been ascribed to autoimmune adrenalitis, which can be isolated (40 %) or in the context of an autoimmune polyendocrinopathy syndrome (60 %) [24]. The clinical symptoms of adrenal insufficiency include weakness, fatigue, anorexia, abdominal pain, weight loss, orthostatic hypotension, salt craving, and characteristic hyperpigmentation of the skin. Regardless of etiology, adrenal insufficiency was an invariably fatal disorder, until the synthesis of cortisone by Kendall and Reichstein in 1949, and the introduction of substitution therapy with life-saving synthetic glucocorticoids. Today, patients with primary adrenal insufficiency are treated with hydrocortisone or cortisone acetate if hydrocortisone is not available, and fludrocortisone to prevent intravascular volume depletion, hyponatremia and hyperkalemia [24]. One of the most important aspects of the management of chronic primary adrenal insufficiency is patient and family education. Patients should understand the reason for life-long replacement therapy, the need to increase the dose of glucocorticoids during minor or major stress and to inject hydrocortisone or methylprednisolone in emergencies.
6.4 Other Disorders Associated with Changes in the HPA Axis
6.4.1 Generalized Glucocorticoid Resistance Syndrome (Chrousos Syndrome)
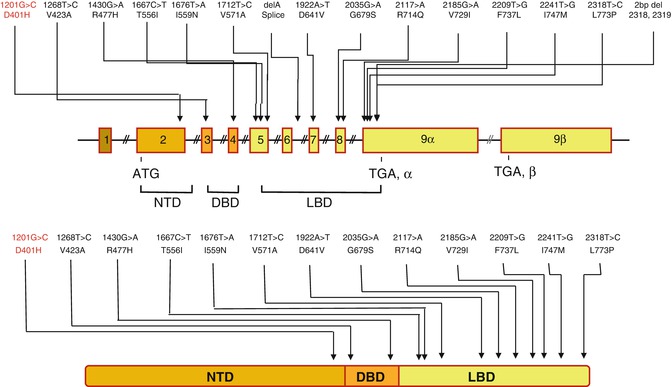
Primary generalized glucocorticoid resistance (Chrousos syndrome) is a rare, familial or sporadic condition, which is characterized by generalized partial end-organ insensitivity to glucocorticoids [42, 43]. Affected patients have compensatory elevation in circulating cortisol and ACTH concentrations, which maintain circadian rhythmicity and appropriate responsiveness to stressors, and resistance of the HPA axis to dexamethasone suppression, but no clinical manifestations suggestive of Cushing syndrome. The clinical spectrum of this condition is broad, ranging from completely asymptomatic cases, displaying biochemical alterations only, to severe forms of mineralocorticoid and/or androgen excess [44]. The molecular basis of Chrousos syndrome has been ascribed to inactivating mutations in the hGR gene [16, 42–45]. Most of these mutations are located in the GR LBD, which alter functions of this subdomain, such as ligand-binding, activation of transcription and nuclear translocation. So far only two mutations have been reported in the DBD of GR: R477H and V423A [46]. All reported pathologic mutations causing familial or sporadic Chrousos syndrome are depicted in Fig. 6.4. Recently, a heterozygous mutation causing glucocorticoid hypersensitivity, ie the mirror image of glucocorticoid resistance, was described [47]. In this patient, Cushingoid features such as obesity and metabolic syndrome were associated with low normal cortisol levels.
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
