Advantages
Disadvantages
References
Maloney-based retrovirus
Self-silencing in pluripotent cells, efficient
Genomic integration, transduces only dividing cells
Takahashi and Yamanaka (2006)
Polycistronic retrovirus
Only single integration event needed
Genomic integration, lower titers/efficiency
Rodriguez-Piza et al. (2010)
HIV-based lentivirus
Transduces dividing and non-dividing cells, efficient
Genomic integration, no silencing in pluripotent state
Yu et al. (2007)
Inducible lentivirus
Temporal control over reprogramming factors
Genomic integration, leaky expression of factors
Brambrink et al. (2008)
Polycistronic lentivirus
Single integration, all factors in one vector
Genomic integration, uneven expression of factors
Chin et al. (2010)
Inducible poly-cistronic lentivirus
Single integration, temporal control of reprogramming
Genomic integration, leaky/uneven expression of factors
Carey et al. (2009)
Integrase-deficient lentivirus
Lower frequency of genomic integration
Lower expression of reprogramming factors
Nightingale et al. (2006)
Excisable lentivirus
Excision of reprogramming factors with minimal genomic footprint
Screening required for multiple excision events, single loxP site left behind
Soldner et al. (2009)
Excisable poly-cistronic lentivirus
Single excision event with minimal genomic footprint
Screening required, uneven expression of factors
Chang et al. (2009)
Adenovirus
No genomic integration
Multiple infections needed, delayed reprogramming
Stadtfeld et al. (2008)
PiggyBac transposon
Complete excision from genome
Still needs secondary screening for excised clones
Kaji et al. (2009)
Inducible PiggyBac transposon
Complete excision, temporal control
Leaky expression, secondary screening
Woltjen et al. (2009)
Transient transfection of DNA plasmids
No genomic integration, no viral vectors
Multiple rounds of transfections needed, low expression of factors, delayed reprogramming
Minicircles
Higher expression than DNA plasmids, integration-free, no viral vectors
Inefficient compared to viral methods, repeated transfections needed
Jia et al. (2010)
Sendaivirus
Non-integrating viral approach; efficient, fast reprogramming
Requires clearance of virus by multiple passaging of iPSC
Small molecules
Transient, dosage controllable, simple methodology
Nonspecific effects/toxicity, reprogramming not yet possible without factors
Huangfu et al. (2008)
RNA transfection
Non-integrating, transient expression of factors, DNA-free approach
Requires multiple transfections, inefficient
Warren et al. (2010)
Protein transfection
Transient, direct delivery of factors, nuclease-free
Multiple transfections needed, inefficient, difficult to reproduce
While generating iPSC has become rote procedure in most laboratories, there is still much variability and need for optimization of reprogramming protocols based on the source tissue/cell type of origin, sample heterogeneity based on patient/disease-specific variation, and reprogramming methodology. iPSC lines generated from even the same cellular origin often display differences in pluripotent characteristics and differentiation potential to various tissues. But still, an iPSC-based approach is proving to be the most viable way to study and treat certain diseases in a patient-specific manner.
iPSCs provide an unlimited source of patient-derived and disease-specific cells, which can then be differentiated into a variety of different cell types. This makes it an ideal model system to study disease pathologies and also to develop new pre-clinical approaches towards therapies. When paired with new, powerful next-generation sequencing and gene-editing technologies, the possibilities are endless for developing patient-specific, personalized medicine-based treatments for genetic and other types of diseases.
Due to the early development of robust protocols for differentiating iPSC to neuronal lineages, their use in modeling neurological disorders has been at the forefront of the iPSC disease-modeling revolution. The earliest iPSC-based disease models were elucidated for inherited neurological disorders such as spinal muscular atrophy (SMA) (Ebert et al. 2009), familial dysautonomia (FD) (Lee et al. 2009), and Rett’s syndrome (Marchetto et al. 2010). In all these cases, the pathology resulting from the disease-causing genetic mutations was elucidated at the cellular and molecular level using iPSC and then suitable drug treatments were discovered based on tests done in these disease-specific iPSC. These tests would otherwise not be possible due to such genetic diseases being rare in occurrence and there being no truly representative model for the disease before the advent of iPSC. The utility of iPSC-based models has been extended beyond inherited genetic diseases to adult-onset or complex neurodegenerative diseases as well. Schizophrenia (SCZD) (Brennand et al. 2011), Parkinson’s disease (PD) (Nguyen et al. 2011), and Alzheimer’s disease (AD) (Yagi et al. 2011) have all been accurately modeled using iPSC, down to the cellular pathology and molecular signature of the disease, and been used to find effective drug treatments against these diseases.
Another organ system that was otherwise very difficult to model was the cardiovascular system, due to lack of access to the human tissue and poor in vitro culturing capability of cells of cardiac origin. iPSC were generated from patients suffering from cardiovascular conditions like long QT syndrome (Moretti et al. 2010; Itzhaki et al. 2011; Yazawa et al. 2011) and LEOPARD syndrome (Carvajal-Vergara et al. 2010) and then converted into cardiomyocytes that could be used for screening for pharmacological agents capable of rescuing the observed electrophysiological defects these cells displayed. Metabolic diseases have also been modeled, for example, iPSC-derived hepatocytes from patients with hypercholesterolemia (Rashid et al. 2010), and insulin-producing cells from Type I diabetic patients (Maehr et al. 2009). Since these initial efforts, a large number of diseases have been modeled successfully using iPSC (Table 7.2).
Table 7.2
A summary of the types of genetic diseases that have been modeled using iPSC. The nature of the disease-specific genetic defect, the terminal cell type derived from the patient iPSC and the associated references are also listed
Disease type | Disease modeled | Genetic defect | Cell type derived | References |
|---|---|---|---|---|
Neurological | Amyotrophic lateral sclerosis (ALS) | Heterozygous L144F mutation in SOD1 | Motor neurons and glial cells | Dimos et al. (2008) |
Neurological | Spinal muscular atrophy (SMA) | Mutations in SMN1 | Neurons and astrocytes | Ebert et al. (2009) |
Neurological | Parkinson’s disease | Mutations in SNCA and LRRK2 | Dopaminergic neurons | |
Neurological | Down’s syndrome | Chromosome 21 trisomy | Teratomas | |
Neurological | Familial dysautonomia | Mutation in IKBKAP | CNS, neurons, hematopoietic, endothelial and endodermal cells | Lee and Studer (2011) |
Neurological | Rett’s syndrome | Heterozygous mutation in MECP2 | Neural progenitor cells | Marchetto et al. (2010) |
Neurological | Schizophrenia | Complex trait | Neurons | Brennand et al. (2011) |
Hematological | Fanconi’s anemia | FAA and FAD2 | Hematopoietic cells | Raya et al. (2009) |
Hematological | β-Thalassemia | Homozygous deletion in β-globin gene | Hematopoietic cells | Ye et al. (2009a) |
Hematological | ADA SCID | Mutation in ADA | None | |
Hematological | Sickle-cell anemia | Homozygous HbS mutation | None | |
Neurological | Adrenoleukodystrophy (ALD) | Mutation in ABCD1 | Oligodendrocytes | Jang et al. (2011) |
Neurological | Huntington’s disease | CAG repeats in huntingtin gene | None | |
Neurological | Fragile X syndrome | Trisomy 21 | None | Urbach et al. (2010) |
Hematological | Polycythaemia vera | Heterozygous V617F mutation in JAK2 | Hematopoietic progenitors | Ye et al. (2009b) |
Hematological | Primary myelofibrosis | Heterozygous mutation in JAK2 | None | Ye et al. (2009b) |
Metabolic | Type 1 diabetes | Multifactorial | β-Cell like cells | Maehr et al. (2009) |
Metabolic | Gaucher’s disease | Mutation in GBA | None | |
Metabolic | α1-Antitrypsin deficiency | Homozygous mutation in α1-Antitrypsin | Hepatocyte-like cells | Rashid et al. (2010) |
Metabolic | Glycogen storage disease | Mutation in glucose-6-phosphate gene | Hepatocyte-like cells | |
Metabolic | Familial hypercholesterolemia | Autosomal dominant mutation in LDLR | Hepatocyte-like cells | Rashid et al. (2010) |
Metabolic | Pompe disease | Knockout of Gaa | Skeletal muscle cells | Kawagoe et al. (2011) |
Metabolic | Hurler syndrome | Genetic defect in IDUA | Hematopoietic cells | Tolar et al. (2011a) |
Cardiovascular | LEOPARD syndrome | Heterozygous mutation in PTPN11 | Cardiomyocytes | Carvajal-Vergara et al. (2010) |
Cardiovascular | Type 1 long QT syndrome | Dominant mutation in KCNQ1 | Cardiomyocytes | Moretti et al. (2010) |
Cardiovascular | Type 2 long QT syndrome | Missense mutation in KCNH2 | Cardiomyocytes | Itzhaki et al. (2011) |
Cardiovascular | Dilated cardiomyopathy (DCM) | Mutation in TNNT2 | Cardiomyocytes | Sun et al. (2012)??? |
Cardiovascular | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | Mutation in PKP2 | Cardiomyocytes | Ma et al. (2013) |
Primary immuno-deficiency | Severe combined immunodeficiency (SCID) | Mutation in RAG1 | None | Pessach et al. (2011) |
Primary immuno-deficiency | Cartilage-hair hypoplasia (CHH) | Mutation in RMRP | None | Pessach et al. (2011) |
Primary immuno-deficiency | Herpes simplex encephalitis (HSE) | Mutation in STAT1 or TLR3 | Mature CNS cell types | Pessach et al. (2011) |
Muscular | Duchenne’s muscular dystrophy | Deletion in dystrophin gene | None | Tchieu et al. (2010) |
Integumentary | Dyskeratosis congenital (DC) | Deletion in DKC1 | None | Agarwal et al. (2010) |
Pulmonary | Cystic fibrosis | Deletion in CFTR | None | Warren et al. (2010) |
CNS | Friedreich’s ataxia (FRDA) | Trinucleotide GAA repeat expansion in FXN | Neurons and cardiomyocytes | Liu et al. (2011) |
Retinal | Retinitis pigmentosa | Mutations in RP9, RP1, PRPH2 or RHO | Retinal and photoreceptors progenitors, RPE cells and rod photoreceptors | Jin et al. (2011) |
Skin | Recessive dystrophic epidermolysis bullosa | Mutation in COL7A1 | Hematopoietic cells, keratinocyte | Tolar et al. (2011b) |
Skin | Scleroderma | Unknown | None | Somers et al. (2010) |
Osteopathic | Osteogenesis imperfect | Mutation in COL1A2 | None | Khan et al. (2010) |
Pulmonary | Pulmonary hypertension (IPAH) | Wnt signaling | Endothelial cells | West et al. (2014) |
Pulmonary | Pulmonary alveolar proteinosis | Mutation in CSF2RA | Monocytes, macrophages |
7.2.1 The Genome Editing Revolution
Recent advances in genome editing technologies have enabled a quantum leap forward in terms of allowing for the unprecedented direct manipulation of the DNA genome of model organisms. Instead of introducing exogenous DNA elements or indirectly targeting gene products by RNA interference to deduce their physiological functions, we can now directly modify genes in situ and knockout genes by targeting their DNA sequence within the nuclear genome. This has been facilitated by a sea-change in the underlying technology, from traditional gene targeting by homologous recombination (HR) (Capecchi 1989), which was inherently very inefficient (targeting only 1 in 106 to 109 cells), to the era of highly precise and efficient genome editing by engineered designer nucleases, which hold immense potential to transform basic science, biotechnology, and medicine.
It had been known for a while that the generation of double-strand breaks (DSB) in the vicinity of the targeted DNA sequence greatly stimulates the efficiency of HR-mediated recombination events (Rudin et al. 1989; Plessis et al. 1992; Rouet et al. 1994). Chandrasegaran and colleagues first targeted specific DNA sequences using zinc finger DNA-binding domains, common to many eukaryotic transcription factors, and linked them to the endonuclease domain of the FokI Type IIS enzyme to create custom-engineered nucleases, called zinc finger nuclease (ZFNs) (Kim et al. 1996). Carroll then co-opted these ZFNs to target genomic sequences and achieve efficient, targeted HR at these sites (Bibikova et al. 2001, 2003). They also showed that in the absence of HR, error-prone non-homologous end joining (NHEJ) at the site of targeted DSBs, such as those generated by ZFNs, could result in deleterious indels that could be used to knockout genes by targeting their genomic sequences (Bibikova et al. 2002). Thus using custom-engineered nucleases, one could achieve highly efficient gene knockout, knock-in, or modification for the very first time.
Efforts had also been undertaken to protein engineer mega-nucleases derived from mobile genetic elements to target custom DNA sequences (Smith et al. 2006); however, this task proved arduous due to the lack of correspondence between their protein residues and their target DNA sequence specificity. ZFNs, on the other hand, could be manipulated to target specific DNA sequences by manipulating the modular assembly of the ZF domains comprising them (Urnov et al. 2005; Miller et al. 2007). However, due to the fact that each ZF domain recognizes a triplet of base pairs, one could not target many DNA sequences which lacked suitable ZFN-targeting specificity, as ZFs targeting all the combination of triplets have still not been engineered. In addition, assembling certain ZF domains adjacently altered their DNA-binding properties in unpredictable ways.
This was largely overcome by the discovery of TALENs, which linked the modular TAL DNA-binding domains found in the plant pathogen Xanthomonas to the FokI endonuclease domain, much like ZFNs (Moscou and Bogdanove 2009; Boch et al. 2009; Christian et al. 2010). Each TAL domain recognizes a single, specific base and occurs naturally as modularly assembled arrays, so TALENs have greater specificity, versatility, and utility for targeting the genome. However, both ZFN and TALEN technologies hit a roadblock en route to more popular and widespread adoption due to the laborious and costly process that assembling such arrays of protein domains entails, as both these technologies rely on protein-based recognition of target DNA sequences.
Given the difficulties of engineering arrays of modular DNA-binding domains, a different mode of DNA recognition would simplify the development of custom nucleases (Hsu et al. 2014). This is where CRISPR (clustered regularly interspersed short palindromic repeats) has revolutionized the field of genome editing by custom-engineered nucleases. A naturally occurring mechanism against viral infection found in a large variety of prokaryotes, the CRISPR system is comprised of a DNA endonuclease, Cas9, whose specificity to target sequences is defined by an RNA component, which binds to target DNA by Watson–Crick base pairing. This unique modality of DNA recognition and binding via a guide RNA (gRNA) of pre-defined sequence makes the engineering of custom CRISPR nucleases as easy as designing an oligonucleotide complementary to the desired target DNA sequence and expressing it as an RNA transcript.
The origins of CRISPR date back to 1987 when Ishino et al (1987) reported the discovery of clustered CRISPR repeats downstream of the iap gene in E. coli (Ishino et al. 1987). As more microbial genomes were sequenced, the presence of similar repeat elements was reported in a large variety of bacteria and archaea and came to be recognized as a unique family of clustered repeat elements (Mojica et al. 2000). These were subsequently named CRISPR and were subdivided into three types of systems (I–III) based on the identity of the cluster of genes found adjacent to these CRISPR loci, which encoded for the protein endonuclease components of the CRISPR system, called the cas proteins (Jansen et al. 2002; Haft et al. 2005). Type I and III CRISPR systems have multiple Cas proteins, but the Type II CRISPR system was comprised of significantly less discrete components and so was the most attractive prospect for development of customized molecular engineering and for adaptation towards targeted genome editing.
Systematic analyses of the CRISPR sequences identified the source of these “spacer” elements to be extrachromosomal in nature, derived from bacteriophage-associated origins (Mojica et al. 2005; Pourcel et al. 2005). This gave rise to the theory that CRISPR systems functioned as an innate immunity-based antiviral defense mechanism. The first experimental proof of CRISPR functioning as an adaptive immune system came in 2007, when the food ingredient company Danisco was studying the dairy production bacterium Streptococcus thermophiles, ostensibly for its use in industrial production of yogurt. They found that the Type II CRISPR system provides antiviral defense by using the spacer elements to dictate target sequence specificity and the Cas proteins to degrade the invasive phage DNA (Barrangou et al. 2007). Subsequently, several salient features of the CRISPR system came to be elucidated; such as the transcription of CRISPR arrays into short crRNA hairpins that guided Cas9 DNA endonuclease activity (Brouns et al. 2008) and the identification of the PAM motif, characteristic of each CRISPR system, that was found to be essential for recognition and cleavage of the target DNA (Bolotin et al. 2005; Deveau et al. 2008). The discovery that the Type II CRISPR system depended on only a single enzymatic component, Cas9, (Garneau et al. 2010) and the fuller understanding of the processing of the crRNA/tracrRNA dual RNA element that guides its endonuclease activity reduced the CRISPR system to its minimal functional components and thus illuminated its potential as a powerful genome editing tool.
The breakthrough studies by Charpentier and Doudna in 2012 first demonstrated the adaptation of the bacterial CRISPR system to target and cleave DNA in vitro, using a single, short gRNA hairpin by incorporating the tracrRNA and crRNA into a single oligonucleotide element (Jinek et al. 2012). This transformed the field of gene targeting, as for the first time a simple and effective methodology was developed to target defined DNA sequences using an RNA-guided, customizable endonuclease. The immense potential of this technology was demonstrated as early as 2013, when the CRISPR system was successfully utilized to achieve efficient and precise genome editing in mammalian cells (Cong et al. 2013; Mali et al. 2013). Since then the CRISPR field has veritably exploded, with new applications and developments being reported in a plethora of model systems.
CRISPR can be used to knockout genes by inducing the error-prone NHEJ repair mechanism to produce deleterious indel mutations within the coding sequences of target genes. By introducing a homologous DNA sequence, DSBs created by CRISPR can be used to modify or knock-in specific DNA sequences at targeted genomic loci by invoking HR-mediated repair. By using a catalytically inactive version of Cas9 (dCas9), which binds to but does not cleave target sequences, one can silence or activate transcription by linking transcriptional repressor or activator domains to the dCas9 protein, or even by interference of transcription by steric hindrance due to the binding of dCas9 (Qi et al. 2013; Maeder et al. 2013; Ran et al. 2013). The use of effector fusions to dCas9 can be utilized for dynamic visualization of genomic loci (Chen et al. 2013) to edit histone modifications in a locus-specific manner (Mendenhall et al. 2013) or to even alter the 3D organization of the genome and its associated chromatin. Using chromatin immunoprecipitation (ChIP), dCas9 could serve as a unique protein tag by which to isolate sequence-specific genomic DNA elements, in the context of its associated chromatin, to study transcription factors, histone modifications, or other chromatin characteristics associated with these genomic loci.
The ability to use CRISPR in multiplexed genome editing by simultaneously introducing gRNAs targeting a variety of genomic sequences is another unique feature of this technology that sets it apart (Cong et al. 2013; Mali et al. 2013). By using tandem gRNAs, one can produce deletions in the genome ranging from a few hundred bases to a few hundreds of thousands of bases (Xiao et al. 2013; Zhou et al. 2014). This multiplexing capability is also taken advantage of in creating a more high-fidelity version of Cas9, by perturbing one of its two DNA endonuclease domains, rendering it into a nickase of double-stranded DNA (Ran et al. 2013). Using a tandem pair of gRNAs offset by an appropriate length of intervening DNA sequence, one can essentially create staggered DSBs in the dsDNA using a pair of nickase molecules, much like ZFNs or TALENs. This improves the specificity of Cas9 by reducing off-target effects, due to the requirement for binding and cleavage by two discrete gRNA recognition elements instead of one.
The ability to easily generate arrays of gRNAs by simply designing unique oligonucleotide sequences against a multitude of genomic targets has been utilized in the generation of genome-wide CRISPR libraries using lentiviral delivery vectors (Wang et al. 2014; Shalem et al. 2014). This allows for genome-wide loss-of-function screens as have been described using RNAi, except now with the ability to actually knock out coding sequences at their genomic loci, and also target other non-coding RNA and regulatory DNA elements. One can link various effector functions to the dCas9 protein to carry out unprecedented transcriptional activation or epigenetic regulation screens at a genome-wide level.
Since its inception, CRISPR genome editing technology has been applied to an exponentially growing repertoire of cellular and animal models (Sander and Joung 2014). It has been used to generate a variety of transgenic animal models from mice to monkeys, where a specific genetic variation can be precisely phenocopied in vivo (Wang et al. 2013; Niu et al. 2014). Even more impressively, transgenic animal models can be generated by directly injecting the Cas9 protein and transcribed gRNA into fertilized zygotes, bypassing the typical ES cells stage for targeting one or multiple alleles, in a much shorter generation time than usual. For mice, novel transgenic models can be derived in a matter of weeks, instead of the traditional time frame of over a year. The ability to use this technology for a variety of organisms is transforming the landscape of experimental biology on almost a day-to-day basis.
CRISPR has already been used to introduce or correct specific mutations in isogenically engineered ESC and iPSC lines (Schwank et al. 2013; Wu et al. 2013). The unique advantage of this type of genome editing in iPSC lies in the fact that one can isolate and culture these cells clonally, and thus derived 100 % pure, isogenic cell lines containing the desired genomic manipulations. These engineered iPSC lines can be differentiated into suitable cell types to observe and study the resultant phenotype. For certain cell types that are otherwise hard to target, one can achieve efficient genome editing at the iPSC stage and then subsequently obtain the desired end cell type by differentiation of these iPSC. One can use CRISPR-mediated genome editing to create true genomic knock-in reporter iPSC lines, where fluorescence, epitope tags, or other detectable markers can be integrated downstream of key genes to monitor and detect their expression. Using such reporter lines, existing differentiation protocols can be vastly improved by detecting and isolating populations of cells expressing the appropriate markers characteristic of the cell types desired at each stage of differentiation. For example, existing lung differentiation protocols would benefit greatly from having reporter lines of genes like Sox2, Nkx2.1, FoxJ1, or FoxA2 to identify various lung progenitor and mature cell types and monitor and isolate these cell populations as they are generated during differentiation (Fig. 7.1).
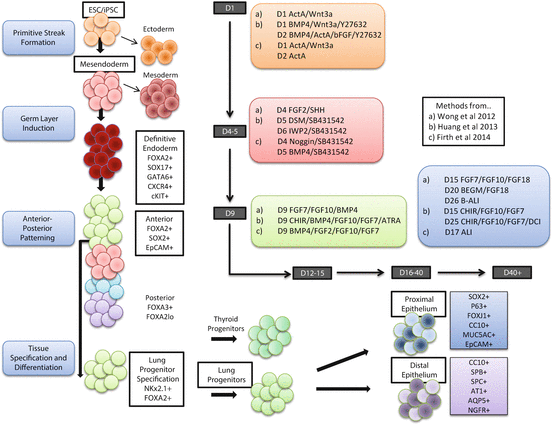
Fig. 7.1
Summary of differentiation from iPSC and hESC to airway epithelium. The diagram highlights the key phases in the development of the lung epithelium from pluripotent stem cells (iPSC/ESC) through definitive endoderm (DE), anterior foregut endoderm (AFE) to lung progenitors (NKx2.1+ and FOXA2+). From here, cells can be matured to generate the proximal conducting epithelium with Club, Ciliated, Goblet and Basal cells and the distal epithelium consisting of variant Club cells, Alveolar type I and II cells. The protocols used by (a) (Wong et al. 2012), (b) (Huang et al. 2013), and (c) (Firth et al. 2014) are included and readers are referred to the original papers and (Gomperts 2014) for more protocol detail
One can already deliver CRISPR as DNA constructs, transcribed RNA, or packaged into viral vectors, like lentivirus or adeno-associated virus (AAV). In addition, inducible control of Cas9 expression will allow better and more precise genome editing. For iPSC, the method of choice is still nucleofection, which allows moderately efficient, but importantly, transient expression of CRISPR for genome editing. Most of the viral approaches, while more efficient, bear the risk of stably integrating a constitutively expressing Cas9 into the cellular genome. This is obviously undesirable due to the possibility of an accumulation of off-target cleavages by a constantly active Cas9, even though given the drastic conformational change in the structure of the Cas9 protein when it binds to gRNA, it seems unlikely it will have any residual activity when not bound to its RNA component (Jinek et al. 2014). Whether Cas9 has the ability to bind naturally occurring RNA transcripts within mammalian cells to catalyze DNA cleavage in a non-specific manner remains to be seen.
Recently, an inducible CRISPR system (iCRISPR) has been described, where TALENs were used to stably knock-in the Cas9 gene into the “safe harbor” AAVS1 genomic locus under a Dox-inducible promoter. This was used to derive human ESC and iPSC lines that can be stimulated to carry out CRISPR-mediated genome editing by Dox-induction of Cas9 expression and transient transfection of target-specific gRNA (Gonzalez et al. 2014). Such inducible expression systems will be invaluable for achieving efficient and precise genome editing, while providing optimal control over the off-target effects and temporal expression of CRISPR. Early reports are emerging of a stably integrated Cas9 mouse model, where one should be able to achieve organ and cell-specific genome editing in vivo by just introducing the gRNA into the appropriate cells, as the Cas9 is ubiquitously and constitutively expressed. As these delivery vectors and expression systems continue to improve, CRISPR technology will break through more biological barriers and technical roadblocks on its path to the conquest of the genome.
7.2.2 Using iPSC to Model the Lung
The seminal work of Yamanaka et al. in 2007 in generating pluripotent stem cells from somatic cells (induced pluripotent stem cells or iPSC) has opened the door for generating patient- or disease-specific pluripotent cells. These have the potential to differentiate to disease-relevant cell types to model disease and screen for novel therapeutic approaches (Takahashi et al. 2007). While iPSC have been widely adopted for differentiating into neural, cardiac, and other cell types discussed above, its implementation in lung biology has been limited. This is largely due to the complexity of lung structure and cell types, which hinders effective functional modeling of the lung in vitro. Therefore, it is necessary to develop cellular models that functionally represent all the different cell types that characterize the different regions of the lung, like the proximal airways, the conducting airways, and the distal alveoli, each of which is represented by a unique combination of cell types and tissue organization.
To be able to utilize iPSC to model lung disease, an effective differentiation protocol is necessary to be able to generate a pseudostratified polarized respiratory epithelium in a dish. In particular, this requires the generation of a specialized post-mitotic multiciliated cell. The differentiation of ESCs and iPSC to airway epithelial cells has recently received increased attention (Wong et al. 2012; Kadzik and Morrisey 2012), and was recently advanced further by Firth et al., 2014, in a study describing the differentiation of human iPSC to a functional respiratory epithelium, demonstrating the generation of multiciliated cells, Clara goblet, and basal cells in a polarized epithelial layer.
Directed differentiation towards endodermal cell lineages like the lung, as well as hepatocytes and pancreatic cells, has been previously attempted by recapitulating the paradigm of endodermal development in the embryo (D’Amour et al. 2006; Gouon-Evans et al. 2006; Green et al. 2011). Activin A can be used to mimic nodal signaling during gastrulation, and thus induce enriched definitive endoderm (DE) from ESC, marked by expression of transcription factors Sox17 and FoxA2 (Gadue et al. 2006; Yasunaga et al. 2005). Addition of Wnt3a, which is required during gastrulation for primitive streak formation (Tam and Loebel 2007), also enhances this process (Nostro and Keller 2012). In order to follow the lung developmental process along the pathway of endodermal maturation, one then has to induce expression of transcription factors Sox2 and FoxA2, characteristic of the anterior foregut endoderm (AFE), while suppressing posterior marker CDX2. Green et al. first described such an endodermal development-mimetic process, by dual inhibition of TGF-β and BMP in ESC-derived DE, by using a pharmacological inhibitor of ActivinA/nodal and TGF-β signaling, along with a physiological inhibitor of BMP, Noggin. This led to increased expression of the foregut marker Sox2 and maintenance of the endodermal marker FoxA2, along with the concomitant suppression of posterior marker CDX2, all indicative of AFE specification (Green et al. 2011).
The AFE gives rise to the pharyngeal endoderm as it develops along the anterio-posterior axis, and consequent dorso-ventral organ patterning gives rise to the lung field, marked by expression of Nkx2.1. Treatment of hPSC-derived AFE with a combination of Wnt3a, KGF, Fgf10, Bmp4, and EGF results in an increase in expression of Nkx2.1, along with a decrease in Sox2 expression, indicating ventralization of the AFE (Green et al. 2011). Following the observation of retinoic acid (RA) signaling in the lung field, but not in the simultaneously developed pharyngeal pouch, Green et al. used RA to increase further the expression of lung markers like Nkx2.1, Nkx2.5, and Pax1, while downregulating pharyngeal cell markers. However, the frequency of Nkx2.1+ FoxA2+ cells was low and specific markers of mature airway epithelium were missing from this protocol.
This protocol was adapted in an Nkx2.1-GFP mouse ESC reporter line by Longmire et al. to purify Nkx2.1+ cell populations from in vitro-derived AFE. The mouse experiments recapitulated the human differentiation protocol (Green et al. 2011) for the most part, except FGF2 was necessary in the ventralization cocktail. They found that the Nkx2.1+ cells sorted from the AFE stage were not entirely mature and also displayed expression of thyroid cell markers. These observations could be due to intrinsic differences between the temporal, dynamic, and physiological characteristics of mouse and human lung development. Addition of a cocktail of 8-bromo-cAMP, dexamethasone, isobutyl-methylxanthine (DCI), KGF, transferrin, and sodium selenite improved distal specification, giving rise to cells expressing mature lung cell markers like surfactant proteins and CC10. Transplantation of sorted Nkx2.1+ AFE cells into decellularized mouse lungs gave sporadic expression of markers found in type I alveolar epithelial cells. Gene expression analysis of these sorted cells had significant overlap with similar cells from the developing mouse lung (Longmire et al. 2012; Gomperts 2014).
Mou et al. also showed that dual TGF-β and BMP inhibition promotes the generation of AFE from DE and drives lung differentiation in favor of neural differentiation. Also, while previous studies generated DE through embryoid body formation, this study differentiated mESC in monolayers using inhibition of TGF-β only (Mou et al. 2012). However, their differentiation only yielded Nkx2.1+ lung progenitors at low efficiencies (10–30 %) and these could only be matured into Nkx2.1+p63+ proximal airway epithelial cells in vivo upon adding KGF and BMP7, inhibiting Wnt and MAPK/ERK signaling and subcutaneous transplantation into mice.
Wong et al. were the first to differentiate human iPSC into mature airway epithelium by using an air–liquid interface (ALI) to mimic the post-natal airway epithelial niche. They were able to obtain higher levels of AFE induction by treatment of hPSC-derived DE with sonic hedgehog (SHH) and FGF2, reporting that 78 % of the resultant cells expressed Nkx2.1 (Wong et al. 2012). However, it should be noted that this high efficiency of Nkx2.1-expressing cells was not reproducible by the efforts of Huang et al. The treatment of these Nk2.1+ cells with FGF10, KGF, FGF18, and low concentration of BMP4, followed by ALI culture, induced proximal lung differentiation, with cells expressing markers of basal, ciliated, and mucous cells, but not club cells, nor any distal lung cells. Functional expression of CFTR was also observed on the apical surface of these iPSC-derived airway epithelia, and this was used as an indicator to compare differentiated lung cells from CF patient-derived iPSC against normal iPSC. The aberrant CFTR expression and function in the patient-derived lung epithelium was rescued using CF corrector compounds. This study elucidated a disease-specific airway epithelial model system, using patient-derived iPSC.
Huang et al. reported a further improvement in the generation of AFE from hPSC, by sequential inhibition of TGF-β/ BMP and then Wnt, to recapitulate developmental anteriorization and ventralization of the DE. This generated Nkx2.1 + FoxA2+ lung progenitor cells with 86 % efficiency, and by following a defined differentiation protocol involving 2 weeks of DCI treatment, these cells yielded over 50 % SP-B+ lung epithelial cells with <5 % SP-C expression (Gomperts 2014). This protocol circumvented the use of ALI culture and appeared to yield a variety of cell types, more biased towards the distal respiratory epithelium, unlike the protocol described by Wong et al., where ALI culture yielded cells more characteristic of proximal airway epithelium (Huang et al. 2014). It has been suggested, due to similarities in their expression profiles, that this protocol by Huang et al. may generate lung epithelial cells that resemble the human fetal lung rather than the adult lung.
More recently, Ghaedi et al. described a protocol that generated alveolar epithelial cells at much higher efficiencies. They reported up to 97 % expression of SP-C, 95 % of mucin, 93 % of SP-B, and 89 % of CD54 in relatively homogenous populations of alveolar epithelium type-II (AEC-II) cells. They also reported that exposure of iPSC-derived AEC-II cells to the Wnt/β-catenin inhibitor, IWR-1, switched the phenotype from that of AEC-II cells to AEC-I cells, with over 90 % expression of type-I markers T-1α and calveolin-1. Under the appropriate culture conditions, iPSC-derived lung progenitors adhered to and repopulated decellularized lung extracellular matrix (Ghaedi et al. 2013). The same group also described the use of ALI culture in a rotating bioreactor for the large-scale production of alveolar epithelial cells for tissue engineering and drug discovery (Ghaedi et al. 2014).
Firth et al. reported the first robust differentiation of iPSC into mature multiciliated respiratory epithelium. iPSC were differentiated via definitive endoderm to AFE, pulmonary endoderm, and then matured in ALI culture to derive a functional pseudostratified polarized epithelium (Firth et al. 2014). Exposure of the basal surface of the pulmonary epithelium to liquid media and the apical surface to air in this ALI culture gave rise to a polarized epithelium, showing the presence of Club cells with CC10 positive vesicles, MUC5A/C positive Goblet cells, and CK5, p63, and PDPN positive basal cells. The basal layer expressing mesenchymal markers was found to be essential for the differentiation and maintenance of the polarized epithelial layer. Primary cilia were evident at the apical surface of the ALI-based epithelium, and when Notch signaling was inhibited, mature multiciliated cells were generated. These were characterized by robust pericentrin staining, indicating the assembly of multiple centrioles at the apical surface, expression of transcription factor FOXJ1, and multiple acetylated tubulin-labeled cilia projections found in individual cells. This iPSC-derived epithelium showed the presence of forskolin-induced chloride currents sensitive to CFTRinh172 in isolated epithelial cells by whole cell patch clamp technique, demonstrating functionality of the epithelial layer (Firth et al. 2014).
Motile multiciliated cells (MCCs) are a population of specialized cells which have exited cell cycle, assembled basal bodies, and project hundreds of motile cilia as they differentiate. Centrioles form the core of the centrosome and are a microtubule-based structure that anchors the cilium (Marshall 2008). The generation of MCC is critical to the function of a respiratory epithelium; their coordinated beating is essential for the movement of mucous and protection of the lung. Inhibition of notch has been shown to be essential for the development of MCC in xenopous, mouse, and human airways (Stubbs et al. 2006, 2012; Rock et al. 2011; Tsao et al. 2011; Marcet et al. 2011; Jurisch-Yaksi et al. 2013). Generation of such cells from pluripotent stem cells, in a Notch-dependent manner, is a major finding by Firth et al., as it provides the opportunity for in-depth study of the development of these cells in the human system and may lead to the discovery of new mechanisms and therapeutic approaches for diseases such as primary ciliary dyskinesia (PCD), which have been difficult to model and understand with the research tools currently available (Noone et al. 2004).
Directed differentiation of iPSC thus provides a renewable source of human airway epithelial cells including multiciliated cells, which can be utilized to study human respiratory diseases that have previously been difficult to study and model in vitro. iPSC provide an unlimited source of cells and also proffer the opportunity for gene editing and clonal expansion of cells for disease modeling. The ability to isolate and culture iPSC clonally is particularly useful for genome editing, as it allows for homogenous gene editing to give rise to isogenic clonal populations of cells bearing the desired engineered genotype. There are several lung diseases with a known genetic origin, such as cystic fibrosis and PCD, which could be corrected by replacement of the defective gene by the correct gene by gene editing technology (Wood et al. 2011). In addition, one can introduce disease-causing mutations to see if they are causative of the pathological phenotype and thus establish de novo models of genetic diseases that are relatively rare in the human population and therefore hard to obtain patient-derived samples to generate iPSC from. It is hoped that patient-specific iPSC cells can be utilized to model the lung, not only to provide a platform for understanding the cellular and molecular mechanisms of respiratory diseases like CF, asthma, and bronchitis among others, but also to generate gene-corrected transplantable cell types capable of engraftment into the lung for direct clinical intervention.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


