(A) A time line showing the major steps of lung development in the mouse and (B) a diagram showing the ontology of cell lineages (bold; with key marker genes indicated), as the endoderm progressively gives rise to the epithelial cells lining the respiratory system. The endoderm and mesoderm germ layers are segregated from a bipotential mesendoderm progenitor during gastrulation at E6.5–7.5. Between E7.5 and E8.5 the endodermal sheet folds over on the anterior and posterior ends to form a primitive gut tube (anterior left) that is patterned along the A–P axis into foregut and hindgut progenitors. At E8.5–9.5 the foregut is further patterned along the A–P and D–V axes to generate organ-specific lineages, with Nkx2-1+ respiratory progenitors (magenta) being detected by E9.0. Between E9.5 and E11.5 the single foregut tube is separated into Sox2+ esophagus and Nkx2-1+ trachea with primary lung buds emerging. The fetal lung grows by branching morphogenesis in the pseudoglandular stage (E12.5–16.5) during which time the epithelium is patterned along the P–D axis into Sox9+ progenitors (green) at the distal tips, which give rise to both Sox2+ proximal airway lineages (purple) as well as distal alveolar epithelium. During the Canalicular (E16.E–17.5) and Saccular (E18–birth) stages of lung development, the proximal epithelia (purple) begins to differentiate into neuroendocrine (n. endo), multiciliated, secretory (club and goblet cell), and basal cells. Alveolarization occurs in the postnatal (P) period, when bipotential alveolar precursors (bp-ATI/II) at the lung periphery differentiate into gas-exchanging ATI cells interspersed with surfactant-expressing ATII cells lining the alveoli (green).
The respiratory epithelial progenitors can first be identified by the localized expression of the homeobox gene Nkx2-1 in a subset of the ventral foregut endoderm at ~E9.0 in the mouse and around 28 days in human gestation (1,5). Morphogenesis of the respiratory system begins between E9.5 and E10.5 when the ventral Nkx2-1+ cells evaginate forming two primary lung buds and the foregut begins to separate into two tubes: a ventral trachea and dorsal esophagus. During the pseudoglandular stage of fetal lung development (E12.5–16.5) the primary lung buds grow through a stereotypical process of branching morphogenesis to generate the highly arborized airway tree. This process is controlled by temporally and spatially dynamic signaling interactions between the growing lung bud tips and the surrounding lung mesenchyme. Branching morphogenesis is tightly coordinated with proximal–distal patterning of the lung to generate proximal epithelial progenitors that give rise to the mucociliary cells of the conducting airways and distal epithelial progenitors that give rise to pneumocytes in the peripheral alveoli. In the canalicular and saccular stages of lung development from E16.5 to postnatal day (P) 5, branching morphogenesis ceases, and the epithelium begins to differentiate as the distal lung walls become thinner and the terminal branches expand forming sacs surrounded by vascular endothelium. Finally in the perinatal period the terminal airway sacs are further subdivided and inflate creating mature alveoli lined by squamous ATI cells that facilitate gas exchange and cuboidal ATII cells that secrete surfactant, allowing the lung to inflate.
The mesenchymal–epithelial interactions regulating lung development are mediated by a number of signaling pathways, including Wnt, bone morphogenesis protein (BMP), TGF-β, fibroblast growth factor(FGF), retinoic acid (RA), and Hedgehog (HH), which are used reiteratively with distinct roles at distinct times during lung organogenesis (1–3). Although research has identified key roles for these factors, precisely how combinatorial signaling is regulated and how temporal and spatial specificity of the cellular responses is controlled are areas of active investigation. This information is critical to effectively mimic lung organogenesis in vitro with stem cells. Here we review the molecular mechanism of lung development, focusing on epithelial differentiation, and highlight some of the important outstanding questions.
Formation and Early A–P Patterning of the Endoderm
In all vertebrate species, signaling by the TGFβ-family ligand Nodal is necessary and sufficient to induce the endoderm and mesoderm germ layers during gastrulation (2). This was first demonstrated when pluripotent cells isolated from pregastrula Xenopus embryos were cultured in high concentrations of Activin (a TGFβ ligand that activates the same receptors as Nodal), which caused them to adopt an endoderm fate, whereas lower Activin concentrations induced the cells to become mesoderm (2). As we describe later, these classic embryological experiments provided the proof of principle for directing endoderm and mesoderm differentiation from mouse and human pluripotent stem cells.
In mice, Nodal is expressed in the primitive streak, an embryonic structure through which cells migrate during gastrulation. As cells pass through the primitive streak and are exposed to Nodal ligands, it is thought that they assume a transient bipotential “mesendoderm” state, expressing genes characteristic of both the endoderm and mesoderm lineages. Cells that emerge from the posterior primitive streak experience lower levels of Nodal and become mesoderm, whereas those that ingress through the anterior end of the primitive streak experience higher Nodal levels and become definitive endoderm (DE) (2). As the DE tissue migrates out of the streak, it intercalates with and displaces the visceral endoderm (VE), an epithelium that gives rise to extraembryonic tissues such as the yolk sac. Nodal-binding to transmembrane receptor complexes results in the phosphorylation and nuclear translocation of the effector protein Smad2, which stimulates the expression of other key transcription factors, including Sox17 and Foxa2 in the endoderm, and Goosecoid and Brachyury (T) in the mesoderm (Figure 1-1) (2). Sox17 is also expressed in the VE, whereas Foxa2 is also expressed in the axial mesoderm; thus coexpression of Sox17 and Foxa2 is a signature of endoderm cells and is commonly used to identify DE cells within stem cell cultures (6,7).
The canonical Wnt/β-catenin pathway cooperates with Nodal signaling to promote specification and prepatterning of the DE during gastrulation. Wnt-binding to Frizzled-LRP coreceptor complexes results in the stabilization of β-catenin, which translocates to the nucleus, where it interacts with Tcf/Lef DNA-binding proteins to regulate transcription. In zebrafish, Xenopus, and mice, β-catenin is essential for gastrulation and helps maintain the high levels of Nodal expression needed for endoderm induction (2). In addition β-catenin/Tcf complexes cooperate with Smad2 to promote the transcription of many mesendoderm genes, including Sox17 and Foxa2 (8,9). Initial prepatterning of the endoderm occurs around the same time as endoderm formation with Foxa2 and Sox17 being differentially required for anterior and posterior endoderm respectively (2).
After gastrulation, between E7.5 and E9.0 in mice, the sheet of DE cells is transformed into a primitive gut tube that is broadly patterned along the A–P axis with the foregut expressing the transcription factors Hhex and Sox2, while the hindgut epithelium expresses the caudal transcription factors Cdx1-4 (Figure 1-1). At this stage in development, regional identity of the endoderm is labile, and if anterior endoderm is experimentally placed in contact with posterior mesoderm, it can be reprogrammed to adopt a hindgut fate (10,11). The posterior mesoderm secretes RA, Wnt, FGF, and BMP ligands, which together promote intestinal identity in the adjacent endoderm. In contrast the anterior region of the embryo expressed a number of Wnt- and BMP-antagonists including Dkk1, Sfrp, Cerberus, Noggin, and Chordin that protect the anterior endoderm from these posteriorizing signals (2). The available evidence suggests that the foregut epithelium is uniquely capable of becoming lung, liver, or pancreas but the molecular basis of this competence is poorly understood.
Specification of Nkx2-1+ Respiratory Progenitors
The foregut endoderm is segregated into organ-specific lineages between E8.0 and E9.5 in mice (Figure 1-1) with specification of the respiratory progenitors defined by the downregulation of Sox2 and the induction of Nkx2-1 in a subset of the ventral foregut cells at E9.0 (Figure 1-2) (5). Although commonly used as a marker of the respiratory lineage, Nkx2-1 is also expressed in the brain and presumptive thyroid and thus is not a completely specific indicator of respiratory identity. Recent expression profiling of the early mouse embryo has revealed a combinatorial transcription factor code for different foregut lineages with expression of Nkx2-1 and the lack of Hhex and Pax8 marking lung, whereas the coexpression of Nkx2-1/Hhex/Pax8 marks the thyroid (12,13). Nkx2-1 directly regulates the expression of many genes in the respiratory epithelium (14), and Nkx2-1 null mutant mice exhibit severe pulmonary defects including trachea-esophageal fistula, poor branching morphogenesis, and a failure in differentiation of the lung epithelium along with thyroid and brain defects (15,16). The fact that Nkx2-1 mutants still make lung tissue indicates that other epithelial transcription factors must act in concert with Nkx2-1 to specify the respiratory lineage. Candidates include a number of Hox-family transcription factors, which display regional expression in the foregut. Consistent with this possibility Hoxa5 and Hoxb5 double mutants exhibit hypoplastic lungs and perinatal lethality (17). An important challenge in the future will be to identify how Nkx2-1, Hox, and other transcription factors interact to control the complete transcriptome of the respiratory lineage.
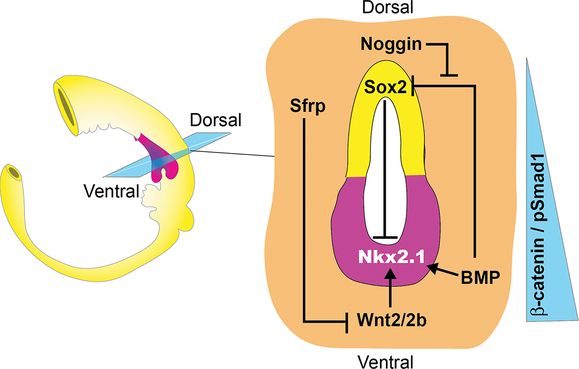
Schematic of an E9.5 mouse gut tube showing a section through the foregut. It shows that Wnt2/2b and BMP paracrine signals from the splanchnic mesoderm (orange) generate a gradient of β-catenin and pSmad1 activity along the D–V axis of the foregut epithelium. Wnt/β-catenin induces Nkx2-1+ respiratory progenitors (pink) in the ventral foregut, whereas BMP/pSmad1 downregulates the ventral expression of the transcription factor Sox2, which suppresses Nkx2-1. Wnt and BMP ligands are counteracted dorsally by the secreted antagonists Noggin and Sfrp, thus maintaining Sox2 expression in the dorsal foregut (yellow) that will give rise to the esophagus.
Experiments with cultured embryonic mouse tissue suggest that between E8.0 and E9.0 dose-dependent FGF signals from the cardiac mesoderm promote the initial segregation of the ventral foregut into lung, liver, and pancreas lineages. High concentrations of recombinant FGF2 induced expression of Nkx2-1 and the ATII gene Sftpc in isolated foregut endoderm, while moderate FGF doses induced liver (18). Similarly, hyperactivation of FGF receptors (FGFR) in Xenopus and chicken embryos expands the Nkx2-1-expressing lung domain (19,20). Pharmacological inhibition in Xenopus embryos indicates that endogenous FGFR signaling via mitogen-activated protein kinase (MAPK) and Akt pathways is required for lung and liver development in vivo and suggests that prolonged FGFR activity is critical for lineage specification, but the molecular mechanisms remain obscure (20,21). This putative role for FGFs in mammalian foregut patterning remains to be genetically validated, as no single or compound Fgf mutant described to date exhibits a failure to specify Nkx2-1+ progenitors, even though a number of FGF ligands play critical roles in fetal lung development (3).
Canonical Wnt signaling is critical for inducing respiratory progenitors. Wnt2 and Wnt2b (Wnt2/2b), expressed in the splanchnic mesoderm surrounding the ventral foregut at E8.5–10.5, are redundantly required for lung development (Figure 1-2). The combined mutation of both Wnt2 and Wnt2b, or the deletion of β-catenin from the epithelium at E9.0, resulted in respiratory agenesis and a lack of robust Nkx2-1 expression, although there may be a very transient lowlevel of Nkx2-1 even in the absence of β-catenin (22,23). In addition, hyperactivation of β-catenin is sufficient to dramatically expand the Nkx2-1 expression domain. Wnt2/2b signaling via β-catenin is also necessary and sufficient to specify respiratory epithelium in Xenopus (24). Epistasis experiments in frog embryos further suggest that FGF-mediated foregut patterning may promote lung fate in part by regulating Wnt2/2b expression in the mesoderm. However, the situation is almost certainly more complicated as compound Wnt2/2b mutant mouse embryos have reduced Fgf10 in the mesenchyme surrounding the nascent lung buds (22). Moreover, analysis of the Fgf10 and Fgf9 mutants indicates that FGFs and Wnts regulate each other’s expression throughout fetal lung development by complex feedback loops both within the mesenchyme and between the mesenchyme and the epithelium (3).
BMP signaling cooperates with Wnt2/2b to promote specification of the respiratory lineage. At E9.0–10, BMP ligands expressed in the cardiac and splanchnic mesenchyme signal to the adjacent ventral foregut epithelium activating the downstream nuclear effectors Smad1/5/8 (Figure 1-2) (25,26). Conditional deletion of BMP receptor genes Bmpr1a and Bmpr1b (Bmpr1a/b) from the endoderm around E9.0, rendering the epithelium unable to respond to BMPs, resulted in reduced Nkx2-1 and tracheal agenesis (27). Curiously Bmpr1a/b deletion also caused ectopic lung buds to emerge from the foregut tube, suggesting that trachea and lung bud progenitors may have different requirements for BMP signaling. Importantly BMPR activity was essential for the ectopic Nkx2-1 expression induced by the experimental hyperactivation of β-catenin. The available evidence suggests that BMP signaling acts in part by repressing Sox2, thereby allowing Nkx2-1 expression in the presumptive lung field (27). Sox2 appears to repress Nkx2-1 transcription, thus downregulation of Sox2 by BMP facilitates Wnt-mediated induction of respiratory progenitors (Figure 1-2). Exactly how Sox2 inhibits Nkx2-1 expression is unclear as a few days later both of these genes are coexpressed in the developing trachea at E11.5 (28), making it unlikely that Sox2 directly represses Nkx2-1 transcription.
Recent evidence from Xenopus embryos suggests that the role of BMPs may be more complex with both “prolung” and “antilung” activities by repressing Sox2 in the epithelium while at the same time restricting the Wnt2/2b expression domain in the mesenchyme (24). This dual function may help to coordinate the location of lung-inducing signals within the region of the foregut competent to respond. In the future it will be important to analyze the enhancers and promoters of the Nkx2-1, Sox2, and Wnt2/2b genomic loci in vivo by chromatin immunoprecipitation (ChIP) to determine how direct DNA-binding of β-catenin/Tcf and Smad1 complexes control their transcription.
D–V Patterning of the Foregut into Trachea and Esophagus
Coincident with induction of Nkx2-1+ respiratory progenitors, differential BMP/Wnt signals also pattern the foregut along the D-V axis such that it separates into distinct tracheal and esophageal tubes. BMP ligands from the ventral mesenchyme are counteracted by Noggin, a BMP-antagonist secreted from the notochord and dorsal foregut (25,26). Similarly the Wnt-antagonists Sfrp1 and Sfrp2 are expressed in the dorsal foregut mesoderm surrounding the developing esophagus, where they appear to restrict the activity of ventrally produced Wnt2/2b (29). This spatial expression pattern of ligands and antagonists results in a graded level of β-catenin and Smad1 activity along the D–V axis of the foregut tube that is highest in the ventral Nkx2-1+ presumptive trachea and lowest in the dorsal Sox2+ presumptive esophagus. The correct balance of BMP/Wnt activity is essential for proper separation of the trachea and esophagus. Reductions in Wnt2/2b or BMP activity, such as in Bmp4–/– or Wnt2–/–;Wnt2b–/– compound mutant mice, results in tracheal atresia (22,25,26), whereas inactivating mutations in Noggin or Sox2 can result in varying degrees of esophageal atresia and trachea-esophageal fistulas (EA/TEF) in mice and human patients (26,28).
The HH pathway is also essential for early respiratory development and separation of trachea and esophagus. Sonic hedgehog ligand (Shh) is expressed throughout the gut tube epithelium, and Shh–/– mice mutant embryos display tracheal atresia with hyperplasic lungs (30), One of the main roles of Shh signals from the epithelium is to maintain the proliferation and survival of the surrounding mesenchyme, which in turn signals back to the endoderm. The combined mutation of Gli2 and Gli3, downstream transcription factors in the HH pathway, results in a more severe phenotype than the Shh mutants, with a complete lack of respiratory system and a single hypoplastic foregut tube (31). Exactly how the Wnt, BMP, and HH pathways modulate cell behaviors to cause the single foregut tube to separate into separate tracheal and esophageal tubes is still poorly understood.
Primary Lung Bud Outgrowth
Initial outgrowth of the primary lung buds around E10.5 in mice requires mesenchymal FGF10 signaling to FGFR2b expressed in the Nkx.1+ foregut epithelium. Fgf10–/– and Fgfr2b–/– knockout mice exhibit a complete lack of lung buds, although respiratory progenitors are specified as indicated by the rudimentary trachea in the mutant embryos (32,33). RA signaling is also essential for primary lung bud formation. If mice lacking the key RA synthesizing enzyme Raldh2 are transiently supplemented with RA between E7.5 and 8.5 to overcome early embryonic lethality, the resulting Raldh2–/– embryos lack lung buds similar to Fgf10 and Fgfr2b mutants (34,35). Studies have shown that RA orchestrates a complex signaling cascade involving FGF, Wnt, and TGFβ pathways to control lung bud growth. On one hand, RA represses expression of the Wnt-antagonist Dkk1 and thus generates a permissive territory where Wnt2/2b can maintain Nkx2-1 expression (36). On the other hand, RA signaling promotes Fgf10 expression by suppressing the TGFβ/pSmad2 pathway (37).
RA may also indirectly maintain Fgf10 and Wnt2/2b expression by promoting the expression of mesenchymal transcription factors such as Tbx4, Tbx5, and Hoxa5. These transcription factors are RA targets in several cellular contexts, and depletion of Tbx4 and/or Tbx5 results in reduced Fgf10 and Wnt2b in chicken and mouse embryos (19,38). Moreover, the Hox cofactors Pbx and Meis have recently been shown to cooperatively regulate Fgf10 transcription (39). It is likely that other signals also cooperate with RA and FGF to regulate lung bud outgrowth. The fact that epithelium-specific deletion of Bmpr1a/b results in ectopic lung buds (27) suggests that BMP signaling may constrain inappropriate lung bud outgrowth, but the relationship of this activity to RA and FGF10 remains to be determined.
The observation that Bmpr1a/b are required for trachea but repress lung budding (27) suggests that trachea and lungs might have distinct progenitors with different molecular programs maintaining their growth. For example, Nkx2-1, Shh, and Bmp4 mutants all have lung tissue but exhibit tracheal dysgenesis (16,25,30), whereas Fgf10 and Fgfr2 mutants lack lungs but form a trachea (32,33). Further work is needed to investigate the segregation of trachea and lung lineages.
Proximal-Distal Patterning, Branching Morphogenesis, and Growth of the Fetal Lung
During the pseudoglandular stage of lung development from E12.5 to E16.5 in mice, the fetal lung grows through stereotypical branching morphogenesis where the distal lung tips undergo a reiterative series of bifurcations to produce the highly arborized tree-like structure of the lung (40). Coincident with branching morphogenesis, the immature respiratory epithelium becomes patterned along the proximal–distal (P–D) axis of the lung. Epithelial progenitors in the proximal regions of the fetal lung, which will line the conducting airways, gives rise to neuroendocrine, secretory, ciliated, and basal cells, whereas the distal peripheral airway epithelium gives rise to gas-exchanging ATI cells and surfactant-producing ATII cells (1,3). Studies have identified a discrete junction between these two epithelial compartments termed the bronchoalveolar duct junction, which displays a clear change in cell type and morphology identifiable around E17 (41). This P–D patterning can first be observed at the molecular level, in the fetal mouse lung by E11 when trachea and proximal lung epithelium re-express Sox2 and the epithelium of the branching distal lung tips express the genes Sox9 and Id2 (Figure 1-3) (1,3). Sox2 itself is required for the formation of secretory and ciliated cells in the proximal airway (28,42) while Sox9 is required for distal alveolar differentiation and normal lung branching (43,44).
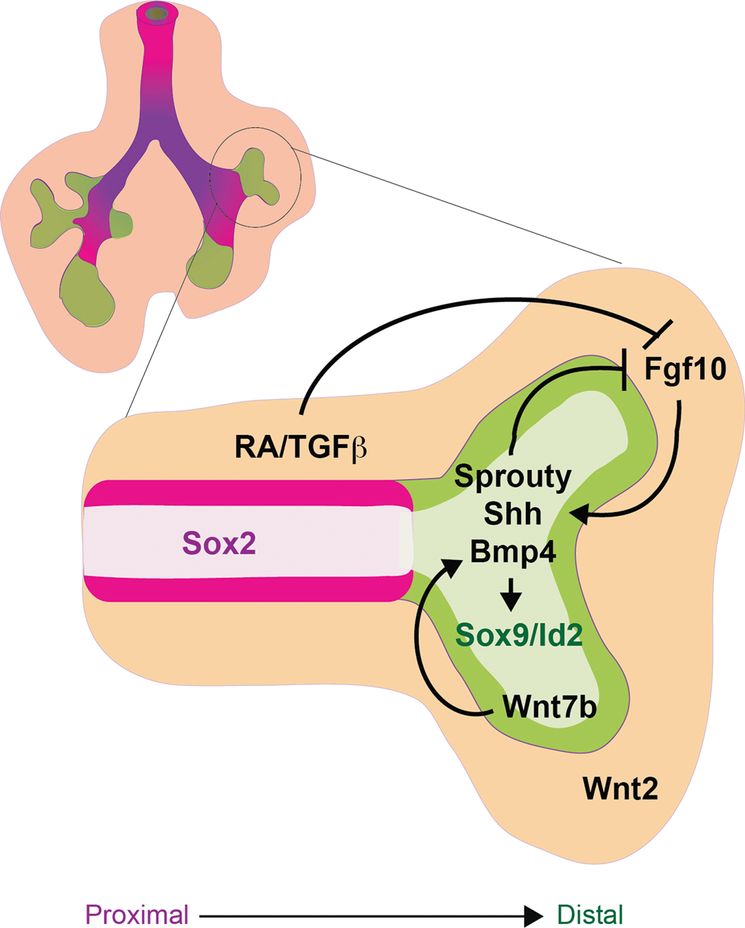
During the pseudoglandular stage of lung development (E12.5–16.5), the fetal lung is patterning along the proximal–distal axis as it grows by branching morphogenesis. The schematic of an early fetal lung with a magnification of one branch shows that the distal tip expressing Sox9 and Id2 (green) is undergoing branching morphogenesis, whereas the proximal airways express Sox2 (magenta). The mesenchyme (orange) surrounding the distal tip expresses FGF10, which along with BMP and Wnt forms a distal signaling center that maintains proliferation and progenitor status of the Sox9/Id2+ distal epithelium. FGF10 induces expression of Sprouty, Shh, and BMP4 in the distal epithelium, which along with RA and TGFβ, signal back through a negative feedback loop to restrict the expression and activity of FGF signaling in the stalk region. As a result of proliferation, epithelial cells that are destined to give rise to conducting airway are eventually displaced from the influence of the distal signaling center, causing them to downregulate Sox9/Id2 and adopt a Sox2+ proximal fate.
P–D patterning, branching morphogenesis, and fetal lung growth are all intimately coordinated by a complex reciprocal cross-talk between the epithelium and mesenchyme involving many of the same signaling factors that regulate respiratory specification and primary lung bud outgrowth namely: FGF, BMP, Wnt, RA, and HH. These interactions generate a signaling center at the distal lung bud tips, which maintains the proliferation of Sox9/Id2+ distal progenitors, promotes branching morphogenesis, and represses Sox2+ proximal fate. As the lung tips grow, epithelial cells that give rise to the stalks of the branching airways eventually become displaced from the influence of distal signaling, and as a result they no longer maintain distal identity and adopt a Sox2+ proximal epithelial fate. Consistent with this model, genetic lineage labeling has shown that Id2-expressing progenitor cells in the distal tips of the E11.5–13.5 fetal lungs can self-renew and contribute to both proximal airway and distal alveolar epithelial lineages (45). Id2-expressing tip cells subsequently lose this multipotent capacity after this developmental stage when they become restricted solely to distal epithelial fates, consistent with a model of increasing lineage restriction of distal progenitors as developmental stages proceed.
Although many of the molecular details remain to be elucidated, a key component of this distal signaling center is the localized expression of FGF10 in the mesenchyme surrounding the distal tips, which signals to Fgfr2b in the epithelium.
Experiments with fetal lung explants and conditional Fgfr2 and Fgf10 knockouts, using CRE-drivers that delete during fetal lung growth (to overcome earlier lung agenesis), revealed that FGF10 is a critical mitogen and chemoattractant for the epithelium that drives the branching morphogenesis program (46,47) and maintains distal Sox9+ cells in a progenitor-like state while repressing proximal Sox2+ airway fate (48,49). Much of FGF10’s activity appears to be mediated by cross-talk with other pathways in the distal signaling center (Figure 1-3). FGF10 promotes the localized expression of Shh, Bmp4, and its target Id2 in the distal epithelium of the branching tips (46). The distal tip epithelium also expresses Wnt7b, which in turn promotes epithelial Bmp4 and Fgfr2 expression (50). Within the epithelium, canonical Wnt/β-catenin activity (stimulated by epithelial Wnt7b and mesenchymal Wnt2) together with autocrine BMP signaling maintains proliferation and differentiation of the distal epithelium, while suppressing proximal fate (Figure 1-3) (1,3). The exact mechanisms by which Wnt/β-catenin and BMP signaling suppresses proximal fate are unclear but it may be similar to the repression of early Sox2 during D–V foregut patterning.
Reciprocal signaling from the epithelium back to the mesenchyme plays a critical role in maintaining the distal signaling center at the growing lung bud tips. In addition to promoting Shh and Bmp4 expression, FGF10 also activates the expression of the FGF-feedback inhibitor Sprouty in the distal tip epithelium. Sprouty then cell-autonomously restricts FGF signal transduction in the epithelium, whereas Shh and Bmp4 signal back to the mesenchyme to suppress the expression and activity of Fgf10. As a result Fgf10 activity is attenuated in the stalk region proximal to the branching buds. This negative feedback along with additional levels of regulation by Fgf9, RA, and TGFβ localizes the dynamic expression of mesenchymal Fgf10 and epithelial Bmp4 to the growing distal tips during branching morphogenesis (Figure 1-3). These complex tissue interactions also regulate the proliferation and differentiation of the mesenchymal components of the lung. How these complex interactions play out in the directed differentiation of stem cell cultures, where endoderm and mesoderm are often present, remains to be fully explored.
Recent studies have discovered a number of additional layers of regulation in fetal lung patterning and differentiation. For example, genetic studies in mice have identified a role for the Hippo/yes-associated protein (YAP) pathway in defining the border between the Sox2+ airway progenitors and the distal Sox9+ presumptive alveolar region (51,52). In addition noncoding microRNAs, which regulate mRNA translation, have been shown to govern the balance between progenitor proliferation and differentiation (53,54), whereas long noncoding RNAs, which are thought to regulate transcription, are reported to modulate early Nkx2-1 expression (55). Epigenetic modification of chromatin can also impact gene expression in the developing lung. For example histone deacetylases (HDACs) have been shown to promote proliferation and P–D differentiation through the regulation of cell cycle genes and Bmp4 expression (53,56). The challenge now is to figure out how these different modes of regulation are integrated and to identify strategies to manipulate them in stem cell differentiation protocols. For a more detailed consideration of the complex signaling interactions that modulate P–D patterning, branching morphogenesis, and fetal lung growth, see recent excellent reviews on the subject (1,3).
Epithelial Differentiation
Differentiation and physiological maturation of the respiratory epithelium occurs in a proximal to distal wave beginning at the late fetal stages and continuing into the postnatal period (1). Starting in the proximal airway, Sox2+ progenitors give rise to neuroendocrine cells (Ascl1+), secretory Club/Clara cells (Scgb1a1+) in mice, and Goblet cells (Muc5ac+) in human, multiciliated cells (Foxj1+) and basal cells (Trp63+) located at the base of the pseudostratified epithelium of the trachea and main stem bronchi (Figure 1-1B). The distal epithelium differentiates last as peripheral lung tips undergo septation and expansion into alveoli lined with gas-exchanging squamous ATI cells (Aqp5+/Pdpn+) that make up more than 90% of the epithelium, interspersed by surfactant-producing cuboidal ATII cells (Sftpc+; Figure 1-1B).
The cell–cell signaling events that govern epithelial differentiation are poorly understood, but gene knockouts in the mouse and in vitro tissue culture experiments have identified many transcription factors important for epithelial differentiation. In some cases these factors are broadly required for the differentiation of many, if not all, epithelial cell types from both the proximal and distal lineages, such as Nkx2-1, Gata6, and Foxa1/2 (1,3,14), whereas others have restricted function such as Sox2 and Sox9 in proximal and distal lineages respectively (28,42–44). However, some transcription factors are critical for specific epithelial cell types such as Ascl1 for neuroendocrine cells, Spedf for goblet cells, Foxj1 and Myb for ciliated cells, and Trp63 for basal cells; for more details on the diverse role of different transcription factors in respiratory epithelium differentiation, we direct the reader to a number of excellent reviews (1,14,53) and references therein.
One signaling pathway known to regulate lineage segregation in the proximal airway is Notch. First Notch signaling between cells within the epithelium defines whether epithelial cells adopt a neuroendocrine (NE) or nonneuroendocrine (Non-NE) fate (57), via the downstream transcription factors Hes1 and Ascl1. Then a second Notch-mediated event regulates the balance between secretory and ciliated cell fate, such that they are distributed in a salt and pepper manner throughout the airway epithelium (58–60). Precisely how the different temporal effects of Notch activation versus Notch inhibition regulates the various lung lineages remains an active area of investigation.
During the postnatal period the alveoli mature to form the gas-exchanging units of the lung, where the differentiating epithelium is in intimate contact with the vascular endothelium and mesenchymal fibroblasts that form the alveolar septa, but how these interactions impact the differentiation of ATI and ATII cells is poorly understood. Recent evidence indicates that glucocorticoids, which are commonly used to treat premature babies, promotes ATII cell maturation (1,41). In vitro studies suggest that transcriptional changes in response to glucocorticoids involve alterations in Nkx2-1 binding and activation of downstream genes (61). Recent single cell transcriptome analysis and lineage tracing studies of the distal lung epithelium during late fetal development have begun to shed new light on a novel bipotential progenitor of the ATI and ATII cells and has identified candidate regulatory proteins that might regulate the segregation of these two lineages (62,63). It appears that these bipotential progenitors express genes characteristic of both ATI and ATII and that lineage restriction involves repression of one of the two genetic programs in late fetal stages, as these bipotential alveolar progenitors are not detected postnatally (62).
Lung Tissue from Pluripotent Stem Cells
Overview
Our understanding of lung development has been significantly advanced through the careful study of animal models, but more work needs to explore the molecular mechanisms driving differentiation of the respiratory epithelium. The goal of this research is to ultimately advance our knowledge of human lung development and disease. A major hurdle to such research is the reality that the developing human lung is generally inaccessible to study. In recent years, the discovery of pluripotent stem cells, including iPSCs, has significantly advanced our ability to model human development and disease in vitro, providing unprecedented access to human cells undergoing developmental cell fate decisions and lung differentiation. Both the discovery of how to generate iPSCs from humans as well as the techniques for differentiating these cells in vitro is founded on many years of basic science investigations into the mechanisms that regulate pluripotency as well as the signaling pathways that enable in vitro differentiation of embryonic stem cells (ESCs) or other pluripotent stem cell populations. In the second part of this chapter we now introduce pluripotent stem cells, including ESCs and iPSCs, before we review the success to date in applying the knowledge gained from studying the molecular mechanisms of respiratory development in animal models to generating lung epithelium from human pluripotent stem cells (hPSCs).
Embryonic Stem Cells
Pluripotent stem cells have the capacity to self-renew indefinitely and form somatic lineages of all three germ layers (ectoderm, mesoderm, and endoderm). Decades prior to the derivation of the first ESC, the concept of a pluripotency emerged from the study of teratocarcinomas. Teratomas (benign) and teratocarcinomas (malignant) are spontaneously occurring tumors that contain derivatives of all three germ layers. Transplantation of a single cell from a teratocarcinoma induced new tumors composed of tissues derived from the different germ layers (64). Teratomas frequently contain areas that resemble early embryos, termed embryoid bodies. This suggested an embryonic nature of the tumor and led to the observation that engrafting pregastrulation mouse embryos at extra-uterine sites of an adult mouse resulted in the formation of a teratocarcinoma (65,66). These observations suggested that a transient population of pluripotent stem cells was transiently present in the epiblast of the developing mouse embryo. Efforts to isolate and propagate these putative pluripotent stem cells from the developing embryo in culture were finally successful in 1981 with the derivation in vitro of pluripotent stem cell lines, termed embryonic stem cells (ESCs), from the inner cell mass of mouse blastocyst embryos (67). Generating mouse ESCs is now commonplace. ESCs have the capacity to form teratomas when injected in vivo and participate in embryogenesis when injected into mouse blastocysts. Transfer of these chimeric blastocysts into foster mouse mothers has demonstrated that ESCs can contribute to all lineages of the developing organism, including the germline, resulting in the production of viable animals. Of note, ESCs are restricted in their ability to contribute to the extra-embryonic lineages such as the trophectoderm. However, when ESCs are injected into a tetraploid blastocyst, the tetraploid host cells can generate the extra-embryonic lineages, while the donor ESCs, remarkably, form the entire fetus proper (68,69). The ability to manipulate the genome of ESCs to target specific genes and subsequently engineer mice (knock-out, knock-in, or knock-down) to interrogate their function has been a powerful development in accelerating our understanding of development and disease (70–72).
Seventeen years after the discovery of mouse ESCs, a method of isolating human ESCs from preimplantation human blastocyst embryos and culturing these cells while maintain their undifferentiated state was first described (73). Similar to mouse ESCs, human ESCs can also differentiate into all three germ layers as evidenced by their ability to form teratomas when injected into mice. Their discovery was met by excitement and controversy. The excitement was fueled by a hope that human ESCs would herald the era of regenerative medicine and the de novo generation of human cells, tissues, and organs. Controversy stemmed from the use of discarded human embryos to generate the ESC lines. Although sufficient discussion of the ethical, political, or religious controversies that followed is beyond the scope of this review, from a scientific perspective ESCs have offered invaluable insights into the genetic program of early human development and have paved the way for the discovery of iPSCs.
Induced Pluripotent Stem Cells (iPSCs)
The discovery of somatic cell reprogramming to produce iPSCs resulted from fifty years of research focused on mechanisms that regulate nuclear reprogramming and the engineered induction of pluripotency in amphibians and mammals (74). In 1962, Jim Gurdon demonstrated that replacing the nucleus of a frog egg with that of an adult frog intestinal cell could produce a viable tadpole (75) and subsequently a viable adult frog. This process of nuclear transfer proved that the nuclei of somatic cells contain the necessary genetic information to generate an entire organism and that the epigenetic and gene expression states that govern and restrict adult cellular identity can be reset or reprogrammed to a pluripotent, embryonic state. In 1997, applying this nuclear reprogramming technique to sheep mammary epithelial cells, Dolly was the first mammal to be cloned (76).
A second field of research that led to the discovery of iPSCs was focused on master transcription factors. Transcription factors (TFs), such as Nkx2-1 reviewed in the first half of this chapter, are diverse and abundant proteins that regulate gene expression by binding to hundreds or thousands of target sequences in the genome. Master transcription factors control cell-specific genes and can potentially determine cell fate. For example, ectopic expression of a single master transcription factor, MyoD, in fibroblasts results in phenotypic conversion into myoblasts (77).
Combining the knowledge of cellular reprogramming, master transcription factor regulation of fate, and the biology of embryonic stem cells, Takahashi and Yamanaka demonstrated in 2006 that the transient, ectopic overexpression of four transcription factors (Oct4, Klf4, Sox2, and c-Myc) could reset the epigenetic state of mouse somatic cells (e.g., fibroblasts) into a pluripotent state virtually indistinguishable from mouse ESCs (78). iPSCs are similar to mouse ESCs in their global gene expression profiles, pluripotency, germ-line competence, and capacity to form an entire mouse (79–81). A year later, this finding was reproduced by reprogramming using human skin fibroblasts into pluripotent cells that phenotypically resemble human ESCs (82,83).
Generation of human iPSCs from skin biopsies, plucked hair follicles, or peripheral blood samples is now well established (82,84,85). Alternative combinations of transcription factors have been identified, including Nanog, Lin28, ESRRB, NR5A2, that establish the core transcriptional circuitry sufficient to reprogram somatic cells into iPSCs (83,86). iPSCs have several advantages over ESCs; they are genetically identical to the person from whom they are generated, and they overcome the ethical controversy that surrounds and limits the study of ESCs or other cell types that require the use of cells obtained from human embryos. As human iPSCs share the capacity for multilineage differentiation, human ESC studies are now focused on recapitulating key developmental processes in vitro, manipulating gene function to interrogate development and disease to generate organ-specific cell types for both pharmaceutical discovery/toxicity studies and potential cell-based therapies.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


