Fig. 5.1
Proposed intra- and inter-cellular signaling mechanisms involved in fibroblast-macrophage crosstalk in chronic non-resolving perivascular inflammation underlying irreversible pulmonary vascular remodeling. In this process, the adventitial fibroblast undergoes epigenetic changes that “lock” this cell into a pro-inflammatory and pro-mitogenic phenotype. Central drivers of this phenotypic arrest are continuously increased NF-κB and HIF signaling, increased PKM2 to PKM1 ratio, increased aerobic glycolysis, suppression of micro-RNAs (mIRs), increased histone deacetylase (HDAC) activity and DNA methylation, and suppressed anti-inflammatory IL-10 generation. The functional consequences are promotion of SMC hypertrophy and proliferation, and activation of resident and recruited macrophages/monocytes into a pro-inflammatory and pro-remodeling phenotype. Activation of the transcription factors STAT3, C/EBPβ, HIF1 alters PKM2 to PKM1 ratio, drives increases in aerobic glycolysis and promotes succinate accumulation. Activation of Arginase1 and decreased IL-10 are additional features of this macrophage phenotype. In a feed-forward loop, pro-inflammatory cytokines and chemokines (IL-6, IL-1, MCP1), metabolites (ATP and lactate), and tissue availability of Arginine and Glutamine, enables the fibroblast-macrophage signaling unit to persist in an activated state and perpetuate vascular remodeling and fibrosis. TIMPs tissue inhibitors of metalloproteinases, MMPs matrix metalloproteinases, SDF-1, stromal cell-derived factor 1, MCP-1 monocyte chemoattractant protein-1, CCR2 C-C chemokine receptor type 2, VWGF vascular endothelial growth factor, HIF1 hypoxia inducible factor 1, HDAC histone deacetylase, STAT3 Signal transducer and activator of transcription 3, PKM1/2 Pyruvate kinase isozymes M1/M2
Role of Extracellular Purine Nucleotides and Adenosine as Regulators of Pulmonary Arterial Inflammation and Remodeling
Extracellular ATP and other nucleotides (ADP, UTP, UDP) and adenosine have long been known as regulators of vascular function, particularly involved in control of blood flow, vascular cell proliferation, migration, chemotaxis, and inflammatory responses [132–134]. Although extracellular nucleotides are the most ubiquitous, they remain the least investigated endogenous signaling molecules. Evidence is accumulating that vascular EC, as well as smooth muscle, epithelial, hematopoietic and other cells, can release ATP in response to hypoxia, inflammation, fluid shear stress, neurotransmission, and other stress-related stimuli and trigger diverse cell-specific responses through metabotropic (P2Y) and ligand-gated (P2X) receptors [134, 135] Most of these environmental stimuli may play a role in controlling extracellular ATP levels in vascular wall adventitia.
A number of studies support the idea that extracellular nucleotides could contribute to the development of vascular disease [136–139]. Extracellular ATP has been implicated in the hyperplasia and hypertrophy of arterial walls in spontaneously hypertensive rats, in regulation of vascular permeability [140, 141], and in control of proliferation and migration of vascular and hematopoietic cells including monocytes [136, 139, 142–144]. Importantly, in stimulating cell proliferation and migration, ATP acts synergistically with cytokines and integrins [136, 143–146] thereby supporting the physiological relevance of extracellular ATP under hypoxic and inflammatory conditions.
Previously, we demonstrated that pulmonary artery adventitial fibroblasts and vasa vasorum endothelial cells (VVECs) are a potent source of extracellular ATP, which acts as an autocrine/paracrine factor augmenting hypoxia-induced VVEC angiogenesis [147, 148]. These angiogenic effects of ATP in VVECs are mediated through P2Y1, and P2Y13 purinergic receptors, prolonged/dramatic activation of PI3K/mTOR and ERK1/2 pathways, and the elevation of cytoplasmic and nucleoplasmic Ca2+ [149]. The responses to extracellular ATP might be particularly important in the hypoxic and inflamed adventitial microenvironment where increased extracellular ATP level can be expected.
In addition to endogenous ATP release, the concentrations of extracellular nucleotides near purinergic receptor are regulated by ecto-nucleoside triphosphate diphospho-hydrolase-1 (NTPDase1/CD39) and ecto-5′-nucleotidase/CD73 [150–152] responsible for extracellular adenosine production from ATP and ADP. However, it is unresolved whether ecto-nucleotidase expression and activities are altered in EC from vessels undergoing hypoxia-induced remodeling. In the vascular system, endothelial E-NTPDase1/CD39 in conjunction with ecto-5’-nucleotidase/CD73, have been implicated in playing a critical role through the termination of pro-thrombotic and pro-inflammatory effects of circulating ATP and ADP and their conversion into adenosine. This keeps the haemostatic process tightly regulated by preventing excessive clot formation and vessel occlusion [152–154]. Data on disordered cellular migration, vascular inflammation, enhanced leakiness, pathological angiogenesis and neointima formation in mice deficient in NTPDase1 (Cd39/Entpd −/−) [150, 155] or ecto-5′-nucleotidase/CD73 [156] demonstrated the importance of coordinated purine homeostasis for proper vascular endothelial functions.
It was demonstrated that significant down-regulation of ecto-nucleotidase activities on vascular endothelium and other cell types during chronic hypoxia and oxidative stress was accompanied by elevated ATP and ADP levels, increased endothelial activation, and concomitant development of vascular disorders [136, 156, 157]. On the other hand, other investigators have shown endothelial nucleotide-inactivating ectoenzymes NTPDase1/CD39 and ecto-5′-nucleotidase/CD73 can be upregulated during acute hypoxia and inflammation. This increases the intravascular adenosine concentrations and dampens excessive inflammatory responses by affecting endothelial barrier function, adhesion and transmigration of lymphoid cells, and expression of other molecules involved in the adhesion cascade [158–160]. Using a direct enzymatic approach, we found that NTPDase1/CD39 and ecto-5′-nucleotidase/CD73 are impaired in VVECs from chronically hypoxic calves, which may serve as an important pre-requisite for consistently elevated ATP and ADP levels at sites of vasa vasorum angiogenesis, enhanced EC proliferation and, eventually, the exacerbation of pathological vascular remodeling [161].
Another important consequence of hypoxia-induced pulmonary vascular remodeling observed in chronically hypoxic animals is infiltration and homing of circulating inflammatory and progenitor cells to the PA adventitia and around the expanding VV, ultimately leading to adventitial inflammation and pathologic vascular remodeling [16, 56]. Although endothelial dysfunction and permeability changes have been intensively investigated in pulmonary artery endothelial cells, the mechanisms that control the pulmonary vasa vasorum permeability remain largely unexplored.
Extracellular adenosine, a product of ATP hydrolysis, has been shown to play a protective role against vascular leak under conditions associated with hypoxia and inflammation [158, 162–166]. Studies from CD73 (−/−) mice provided evidence that extracellular adenosine reverses hypoxia-induced vascular leakage in different organs, especially in the lung [165]. Adenosine binds to A2A and A2B receptors that function by activating adenylyl cyclase and generation of cAMP, or to A1 and A3 receptors that function by inhibiting adenylyl cyclase and increasing intracellular Ca2+ levels by a pathway involving phospholipase C [135, 165, 167–169]. Previous studies demonstrated a protective role of A2B adenosine receptors in hypoxia-induced vascular leak in adenosine receptor-knockout mice [164, 165]. Consistent with this observation, a recent report indicated permeability of pulmonary artery endothelial cells is regulated by A2A and A2B adenosine receptors and an adenosine transporter, pointing out an importance of both the extracellular and intracellular adenosine [170]. However the role of individual adenosine receptor subtypes in vasa vasorum permeability was unknown.
In agreement with previous findings, our recent study revealed a potent barrier protective effect of extracellular adenosine on the VVEC. The response was observed in VVEC isolated from both control and chronically hypoxic animals, but the cells from control animals exhibited more prominent response. Using pharmacological and genetic approaches, we found that the effect of adenosine on VVEC permeability is mediated mostly by A1R, while A2AR, A2BR and A3R are not likely to be involved. Importantly, a decrease in expression of A1R in VVEC of hypoxic animals correlates with a lower Trans Endothelial Resistance (TER) compared to VVEC of controls. The evidence of A1R involvement in barrier protection is also consistent with an anti-inflammatory role of A1R in several tissues [171–173] and may explain both anti-inflammatory and barrier-protective functions of A1R in vasa vasorum. For example, recent studies reported that A1R in lung microvascular endothelial cells participates in microvascular permeability and leukocyte transmigration [173] and in anti-inflammatory preconditioning [174]. Data from animal models also indicate the involvement of A1R in attenuation of endotoxin-induced lung injury, pulmonary edema, and alveolar destruction. Activation of adenosine A1 and A2 receptors have also been shown to reduce endotoxin-induced cellular energy depletion and edema formation in the lung [175]. However, our findings are different from the results in human lung microvascular endothelial cells, which demonstrated a role of A2AR in adenosine-induced barrier enhancement [175–177]. In contrast to studies showing cAMP-dependent regulation of endothelial barrier function, our studies revealed a role of Gαi/PI3K/Akt pathway actin cytoskeleton remodeling in mediating the effects of A1R activation, suggesting a non-canonical (possibly cAMP–independent) pathway of VVEC barrier regulation [178].
TNF-α and Vasa Vasorum Permeability
TNF-α, one of the most potent pro-inflammatory factors, regulates vascular endothelial cell permeability through stress fiber formation and interruption of cellular junctions [179–181]. TNF-α expression level and activity can be up-regulated under hypoxia, inflammation, and PH [182–185]. It has been shown that among several cell types, macrophages and perivascular adipocytes are potent sources of TNF-α [185, 186]. As the presence of macrophages was observed in pulmonary artery adventitia of chronically hypoxic animals [16], it can be expected that TNF-α, may have a paracrine effect on adventitial vasa vasorum in the pulmonary artery wall. The data from our recent study also show that TNF-α decrease the TER in control VVECs (VVEC-Co), and this effect of TNF-α was blunted by adenosine [178]. Interestingly, TNF-α failed to decrease TER in VVEC isolated from hypoxic animals. This suggests a possibility of persistent phenotypical changes in VVEC in response to chronic hypoxia that could involve TNF-α and adenosine receptors, as well as components of intracellular signaling pathways. We demonstrated that TNF-α was unable to further impair barrier function in VVEC-Hyp, (contrary to VVEC-Co), suggesting that exposure of VVEC to chronic hypoxia impairs these cells’ permeability. Finally, we showed a significant attenuation of TNF-α-induced VVEC permeability upon adenosine treatment, indicative of the barrier-protective effect of adenosine.
In a view of pathologic consequence of hypoxia-induced vasa vasorum neovascularization and its function as a conduit for circulating inflammatory cells to the vascular wall, our data indicate that down-regulation of A1R in chronic hypoxia may represent a pathological mechanism of dysregulation of vasa vasorum barrier function. This may lead to pulmonary vascular remodeling and inflammation, such as that observed in hypoxic PH. We propose that A1Rs can be recognized as a vascular bed-specific and novel therapeutic target to regulate vasa vasorum barrier function and pathologic vascular remodeling in chronic hypoxia.
Regulation of Inflammatory Cells in Adventitia by Extracellular Nucleotides and Adenosine
As mentioned above, hypoxia-induced pulmonary vascular remodeling observed in chronically hypoxic animals is accompanied by extravasation and homing of circulating inflammatory and progenitor cells to the PA adventitia and around the expanding VV network [16, 56]. A number of studies have demonstrated that monocyte/macrophage inflammation functions are tightly regulated by extracellular nucleotides and adenosine. Extracellular ATP exerts pro-inflammatory and cytotoxic effects on monocytes/macrophages via regulation of cytokine and chemokine production including IL-1α, IL-6, IL-18, and TNFα, αϑδ eicosanoids, including leukotrienes LTB4 and LTC4 and prostaglandins PGE2 and PGD2 [132]. Therefore, it is expected that in the adventitial microenvironment, extracellular ATP endogenously released by adventitial fibroblasts and vasa vasorum in response to local hypoxia and oxidative stress, functions as an inflammatory mediator via activation of macrophages and another type of inflammatory cells. In contrast, extracellular adenosine may prevent excessive accumulation and the activation of inflammatory cells in the adventitia via modulation of cytokine production and inhibition of chemotactic response to ATP [132] (Fig. 5.2).
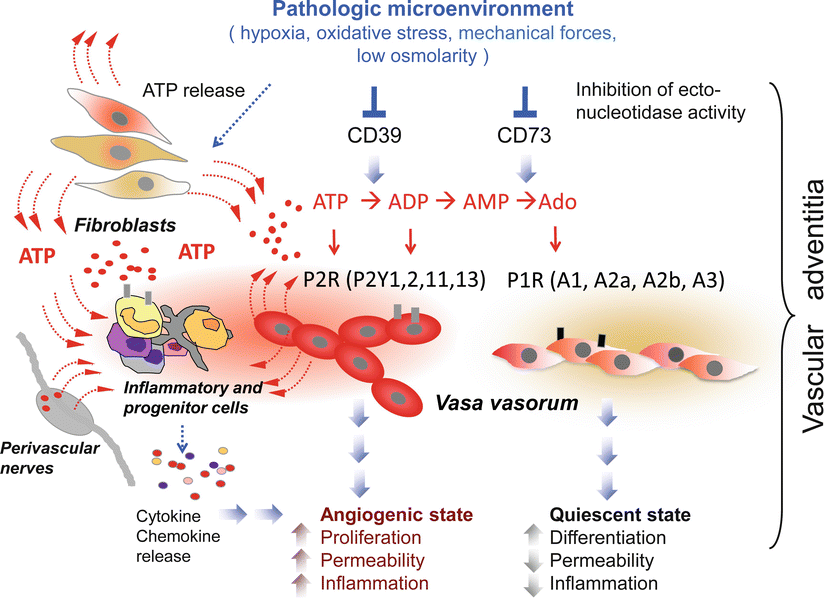
Fig. 5.2
Schematic view of the role of extracellular nucleotides in pulmonary artery adventitia. Stress conditions such as hypoxia, inflammation, oxidative stress, and mechanical forces stimulate ATP release from adventitial fibroblasts, vasa vasorum endothelial cells, perivascular nerves, and inflammatory cells. In addition, extracellular ATP stimulates monocytes/macrophages to release multiple inflammatory mediators that creates a pathologic microenvironment in the pulmonary artery adventitia. Endogenously released ATP, by acting on P2 purinergic receptors (P2R) results in angiogenic activation of the vasa vasorum, characterized by increased proliferation and dysregulated barrier properties. Elevated extracellular ATP is subsequently hydrolyzed by ecto-enzymes, NTPDase1/CD39 to ADP and AMP and by ecto-5′-nucleotidase/CD73 to Adenosine (Ado). In turn, extracellular adenosine by acting on P1 purinergic receptors (P1R) induces a phenotypic switch of the vasa vasorum endothelial cell to a more quiescent state, characterized by low proliferation rate and improved barrier function. Inhibition of NTPDase1/CD39 and ecto-5′-nucleotidase/CD73 activities by hypoxia and oxidative stress results in consistently elevated levels of extracellular ATP and lower levels of adenosine that eventually exacerbate pathological vascular remodeling
Chronic Hypoxia, Inflammation, and Pulmonary Vascular Function: Relationship to Oxidative Stress
To fully understand the impact of chronic hypoxia and unresolved inflammation on the pathogenesis of pulmonary vascular disease, it is essential to also recognize the relationship of inflammation to oxidative stress and altered redox regulated signaling. Accumulating evidence indicates that reactive oxygen species (ROS) are increased in PH, including chronic hypoxia-induced PH, contributing to both vasoconstriction and pulmonary vascular remodeling [187–193]. ROS are generated as a result of inflammation, and conversely, ROS activate inflammatory pathways, thus both processes are closely linked and central to pulmonary vascular dysfunction.
The primary ROS implicated in cell signaling in the vasculature are superoxide and hydrogen peroxide. Superoxide is rapidly catalyzed to hydrogen peroxide and oxygen by the superoxide dismutase (SOD) family of antioxidant enzymes, or, in the face of inadequate SOD, reacts with nitric oxide, inactivating its bioactivity and forming toxic products like peroxynitrite. Hydrogen peroxide has been implicated in both normal cell signaling and pathologic signaling. It is scavenged by antioxidant enzymes including catalase and glutathione peroxidases, or in the presence of iron, can form the highly reactive hydroxyl radical.
The main sources of superoxide and hydrogen peroxide in the vasculature are the NAPDH oxidases, uncoupled eNOS, mitochondrial electron transport chain, and xanthine oxidase [194, 195]. The NADPH oxidases are a family of transmembrane multimeric proteins that are comprised of 7 different homologs with various regulatory subunits; Nox1, Nox2, Nox4 and Nox5 are expressed in the pulmonary vasculature. The Nox2 isoform of NADPH oxidase is abundantly expressed and induced in inflammatory cells present in the vessel wall, in particular in the PA adventitial compartment. In addition, the PA adventitial fibroblasts contain predominantly Nox4, as well as Nox2, and are a major source of ROS contributing to the “outside-in” effects of the PA adventitia on pulmonary vasoconstriction and remodeling [12, 196, 197]. Nox2 and Nox4 are also expressed in endothelial cells and PASMC, and collectively, the NADPH oxidases have been shown to promote vascular dysfunction. While endothelial nitric oxide synthase (eNOS) generates nitric oxide under physiologic conditions, in disease states, the two enzymatic domains of eNOS become uncoupled, resulting in release of superoxide. The uncoupling of eNOS can occur with deficiencies in l-arginine substrate; increased ADMA, an l-arginine analog; increased arginase activity; or BH4 deficiency due to low production or oxidation. The mitochondrial electron transport chain is also well established to be a major source of ROS generation and numerous studies implicate mitochondrial dysfunction and mitochondrial-derived ROS in pulmonary vascular disease [198–200]. However, the role of the mitochondria in pulmonary vascular disease is complicated by the opposing observations that mitochondrial ROS production decreases in fawn hooded rats with spontaneous PH and mitochondrial ROS are decreased in the setting of hypoxia. Using new tools, there is evidence that ROS production may even be regulated differently in the mitochondrial matrix compared to the mitochondrial intermembrane space [195, 201]. Xanthine dehydrogenase can be oxidized to xanthine oxidase (XO, also known as xanthine oxidoreductase, XOR), which then catalyzes the conversion of hypoxanthine and xanthine to uric acid with concurrent release of superoxide. XO has been shown to be an important source of oxidative stress in hypoxia induced PH and the lamb model of PH associated with elevated pulmonary blood flow [195, 202–205]. Furthermore, XO-derived ROS contribute to injury in a number of processes associated with inflammation including ischemia-reperfusion injury, acute lung injury, COPD and cigarette exposure, and cancer [188, 196, 205–216]. Our recent work demonstrates that XO promotes the inflammatory state of pulmonary mononuclear phagocytes through effects on HIF-1α, further supporting the close link between ROS and inflammation [217]. The production of ROS by these important sources can be activated by pro-inflammatory cytokines, including interleukins and tumor necrosis factor-α, and conversely, the ROS generated in the vessel wall can augment inflammation by activating redox sensitive targets including key transcription factors, NF-κβ, AP-1, and HIF. In addition, ROS can modulate a wide range of other signaling molecules that impact inflammation, proliferation, migration, differentiation, and matrix production [194]. The best described redox sensitive targets include receptor and non-receptor tyrosine kinases, serine/threonine kinases, protein tyrosine phosphatases, growth factors, adhesion molecules and ion channels [194–196, 218].
The importance of ROS in inflammation and pulmonary vascular dysfunction is further evidenced by the critical role of antioxidant enzymes. The major vascular antioxidant enzymes are SOD, catalase and glutathione peroxidase [195]. The extracellular isoform of SOD, extracellular superoxide dismutase (EC-SOD or SOD3) is the dominant SOD isoform in the vasculature and is highly localized to the vascular adventitia [219, 220]. In multiple models of vascular disease, including chronic hypoxic PH, loss of SOD3 enhances inflammation and vascular dysfunction, while overexpression of SOD3 protects [194, 221–223]. Mice lacking SOD1 also exhibit exaggerated chronic hypoxic PH [224] and SOD2 has been shown to be decreased in the lungs of fawn hooded rats, and the pulmonary arteries of patients with IPAH [225–229]. Though catalase or glutathione peroxidase have not been shown to be consistently altered in PH, loss of catalase can lead to hydrogen peroxide mediated inactivation of SOD3 and glutathione peroxidase may increase to compensate for oxidative stress but remain overall insufficient [195, 230].
Collectively, these studies strongly support the premise that generation of ROS contribute to the chronic inflammation and pulmonary vascular dysfunction in the setting of hypoxia by further augmenting inflammation as well as other redox sensitive signaling pathways involved in disease pathogenesis (Fig. 5.3).
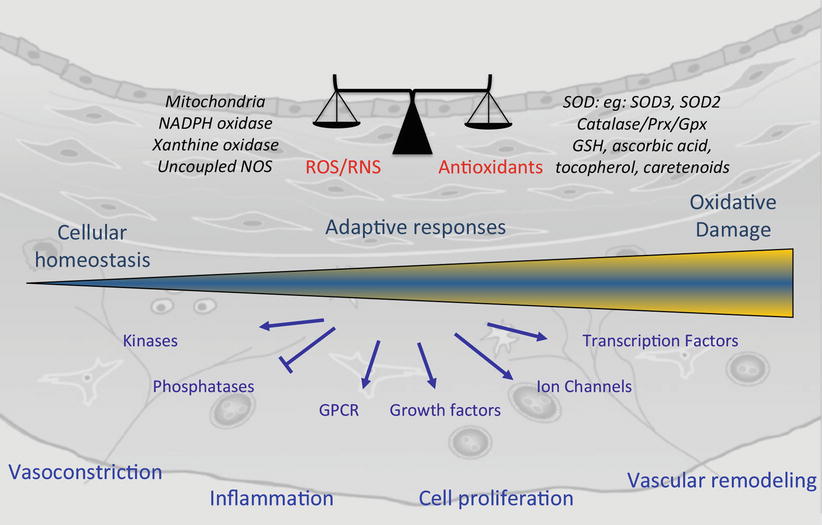
Fig. 5.3
Redox regulated signaling in pulmonary vascular remodeling. ROS/RNS are important in cellular homeostasis and their levels are tightly controlled by enzymatic and non enzymatic antioxidant systems. In the setting of disease, an increase in ROS due to increased production or decreased antioxidant defenses may initially enable adaptive responses but eventually leads to oxidative stress. ROS can directly activate key redox sensitive targets that lead ultimately to pulmonary vasoconstriction, inflammation, cell proliferation and vascular remodeling. ROS reactive oxygen species, RNS reactive nitrogen species, NADPH nicotinamide adenine dinucleotide phosphate, NOS nitric oxide synthase, SOD superoxide dismutase, SOD3 extracellular SOD, SOD2 mitochondrial SOD, Prx peroxireductases, Gpx glutathione peroxidase, GSH glutathione, GPCR G-protein coupled receptor
Contribution of Epigenetics to Chronic Hypoxia-Induced Lung Vascular Inflammation and Remodeling in PH
It is increasingly appreciated that chronic inflammatory microenvironments lead to stable heritable changes in gene expression and cell function without modification of the underlying DNA base composition; i.e. epigenetic change [231, 232]. There are at least three distinct mechanisms of epigenetic regulation, DNA methylation, histone modifications, and gene silencing, mediated by microRNAs (miRs). These pathways of gene regulation are often altered in human diseases such as cancer and are well recognized to contribute to uncontrolled cell growth, migration, and invasion [233–235]. Further, it is also important to note that there are substantial interactions between these epigenetic pathways involved in gene regulation. Studies in a wide variety of cells, mostly cancerous, have demonstrated that hypoxia can regulate changes in all of these epigenetic regulatory pathways [236, 237].
Changes in epigenetic modifications have recently been associated with PH, a disease characterized not only by chronic inflammation but also by mesenchymal cell (SMC and fibroblast) proliferation, resistance to apoptosis, and fibrosis as noted above [15, 238]. For instance, recent studies have demonstrated that superoxide dismutase (SOD) 2 expression is decreased in pulmonary arteries and plexiform lesions because of hypermethylation of CpG islands in the SOD2 gene [227]. Reversal of the methylation was shown to rescue SOD2 expression and to inhibit proliferation and to increase cell apoptosis of PASMCs from the Fawn Hooded rat [227]. Histone acetylation has also been shown to play an important role in the development of PH, and specifically hypoxic PH. Increased HDAC expression has been reported in lung tissues of patients with IPAH as well as in tissues from hypoxia induced PH rats and calves [239]. Specific increases in Class I HDACs were observed in the fibroblast from hypoxic animals as well as in cells from IPAH patients. Treatment with Class I HDAC inhibitors markedly decreased cytokine/chemokine mRNA expression levels in fibroblasts as well as in their ability to induce monocyte migration and pro-inflammatory activation. Most interestingly studies with several HDAC inhibitors, including the Class I specific HDAC inhibitor were shown to both suppress and reverse hypoxia induced cardiopulmonary remodeling in rats [88, 239].
Increases in HDAC expression, and thus histone acetylation, also were shown to contribute to the abnormalities of mesenchymal cell proliferation in rats, calves, and sheep [26, 88, 239, 240]. Studies in cells derived from the hypertensive pulmonary circulation of all three species have demonstrated that HDAC inhibition results in a decrease in proliferation mediated in part because regulation cell cycle regulatory genes, including p16INK, p21, and p27. In PASMCs of hypoxic sheep fetuses, HDAC inhibition also decreased PDGF-induced cell migration and ERK activation as well as modulating global DNA methylation, again consistent with the idea that interactions among the epigenetic mechanisms are important in controlling cell phenotypes [239, 240]. It is also interesting to note that chromatin immunoprecipitation analysis experiments have shown that TGF-β which is thought to play an important role in hypoxic and other forms of PH, increases binding of Smad2/3, Smad4, and the transcriptional co-repressor HDAC-1 to the PPAR-γ promoter. This reduces PPAR-γ, which has been shown to be associated with heightened proliferation and other abnormal activities in cells from the PH animals. Treatment with the PPAR-γ agonist, rosiglitazone, prevented this interaction again implicating the role of HDACs in chronic hypoxia induced remodeling [241].
Recent studies have also implicated miRs in the development of PH. miR204 expression has been shown to be decreased in animal models of PH and in human patient samples and rescue of miR204 reverses PH in rats [242]. miR17 has also been shown to be upregulated in hypoxia and monocrotaline-induced PH and inhibition of miR17 improved mitigated PH in both species [243]. miR17 has also been shown to be upregulated by hypoxia in cultured human PASMCs. In addition, it was shown that inhibiting miR17-5P expression decreased hypoxia-induced arginase protein levels in human PASMCs, which has been shown to be involved in promoting proliferation [244]. Several other miRs and miR targets have been shown to be involved in the development of hypoxic PH. Studies by Gou et al. found that miR210 is the predominant miR induced by hypoxia in human PASMCs [245]. Others have called miR210 the master hypoxamir [246]. Transcriptional induction of miR210 is HIF1α dependent. Inhibition of miR210 in human PASMCs causes significant decrease in cell number under hypoxic conditions due to increased apoptosis probably via regulation of the transcription factor E2F3 [245]. Another interesting study has demonstrated that hypoxia and miR210 increase proliferation in idiopathic pulmonary fibrosis (IPF) fibroblasts [247]. miR210 expression markedly increases in IPF fibroblasts in response to hypoxia and knockdown of miR210 decreases hypoxia induced IPF-fibroblast proliferation. Importantly, the investigators showed that silencing HIF2α inhibits the hypoxia mediated increase in miR210 expression, indicating that in certain cells HIF2 is upstream of miR210 [247]. Importantly, in situ analysis of IPF lung tissue demonstrated that miR210 expression was distributed similarly with HIF2α and the hypoxic marker CA-1X in cells within the IPF fibrotic reticulum. Thus, the authors raised the possibility that a pathologic feed-forward loop could exist in fibrotic lungs in which hypoxia promotes fibroblast proliferation via stimulation of miR210 expression, which in turn worsens hypoxia. Other miRs including miR-145, 21, and 206 have been implicated in hypoxic forms of PH [248–251]. miR 21 expression is increased in the distal PA’s of hypoxia exposed mice and putative targets of miR 21 including BMPR2 were increased [252]. Sequestration of miR21 diminished chronic hypoxia-induced PH and vascular remodeling. miR145 is also of particular interest as it was shown that miR145 was increased in hypoxic mouse lungs and that miR145 deficiency (KO mice) and anti-miR145 both resulted in significant protection from hypoxia-induced PH.
Another recent study has demonstrated the complex relationship between gene regulation and gene expression with regard to epigenetic mechanisms. Wang et al. demonstrated in adventitial fibroblasts from neonatal calves with severe hypoxia induced PH that miR124 expression was decreased and that miR124 directly regulated MCP1 expression and indirectly regulated proliferation through the alternative splicing factor PTBP1 [26]. Further, it was shown that down-regulation of miR124 was mediated through Class I specific HDACs. The authors discovered that treatment of PH fibroblasts with HDAC inhibitors, including SAHA, apicidin, and OSU42 led to significant increases in miR124 while decreasing direct targets of miR124, including the alternative splicing factor PTBP1 and the pro-inflammatory cytokine MCP1. Thus, miR expression itself, under hypoxic conditions, is regulated by epigenetic modifications, specifically the removal of acetylation marks on histone resulting in more condensed chromatin structure and inhibition of transcription. Collectively, these epigenetic changes, which occur in the setting of hypoxia and inflammation, begin to explain the constitutively activated phenotype of PH fibroblasts [26].
The aforementioned studies demonstrate that, while positive adaptive responses to acute hypoxia in the lung are probably crucial for maintaining homeostatic responses, long-term chronic hypoxia can result in responses that are detrimental to the lung and the lung vasculature. The classical adaptive responses to hypoxia, that aim to restore oxygen homeostasis in tissues, including the lung, are regulated by the HIF family of proteins. It is thus not surprising that in addition to work specifically in the lung that there is much current research, which implicates epigenetic mechanisms in modulating the cellular response to hypoxic environments [237, 253, 254]. There is increasing evidence supporting the idea that the activity of hypoxia induced transcription factors, including HIF is superimposed on a background of epigenetic changes that are essential for determining the cellular or tissues specific hypoxic response. For instance, interesting work demonstrates that epigenetic modifications at the DNA and histone level have the ability to dictate HIF binding to target gene promoters and thus to regulating hypoxic gene expression. Further, hypoxia itself is a potent inducer of chromatin remodeling via the regulation of enzymes that modulate DNA methylation and histone modifications. Long-term adaptation to chronic hypoxia involves significant modification of chromatin structure in order to maintain the hypoxic phenotype, even in the absence of HIF1. It is important to note that data in the pulmonary circulation, along with many other organs, suggest that chronic hypoxia is capable of inducing changes in gene expression that are independent of classical HIF pathway. Again, this is probably due to alterations in the methylation status of gene sequence or modification of the histone code, which are likely mediated through prolonged alterations in epigenetic modifying enzymes.
There are four current opinions on the interactions of epigenetics and hypoxia:
1.
HIF stabilization is influenced by the epigenetically controlled expression of Von Hippel Lindau (VHL) and PHD3.
2.
Epigenetic mechanisms regulate HIF binding by maintaining a transcriptionally active chromatin confirmation within and around HIF binding site regions. This may occur through the action of the HIF1α co-activation complex or through direct modifications of HR rebinding sites, which prevent HIF binding.
3.
A significant number of histone dimethylase enzymes are direct HIF1 target genes and therefore play a role in the regulation of transcription during hypoxic responses.
4.
Significant global changes in histone modifications and DNA methylation occur in response to hypoxic exposure.
Investigation of all these possibilities in the setting of chronic hypoxia in the lung will be important as it will likely dictate new therapeutic approaches to ameliorate chronic hypoxia induced lung tissue responses. At present, current data points to the possibility that HDAC inhibitors are important in controlling hypoxic generated proliferative inflammatory and fibrotic responses. Clearly, other possibilities that will be aimed at DNA methylation and/or histone methylation will be tested in the not too distant future.
Conclusion
Hypoxic, or WHO Group 3, PH comprises a heterogeneous group of diseases sharing the common feature of chronic hypoxia-induced pulmonary vascular remodeling, which is usually mild to moderate in nature and which can be largely reversible as compared with the progressive irreversible disease seen in WHO Group I disease. Despite the fact that patients with WHO Group 3 PH are much more likely to die as a result of their underlying lung disease than from complications of PH, the presence of PH in these patients is the most important marker of morbidity and mortality. Furthermore, this patient population is exponentially larger than that of patients with PAH, yet numerous clinical trials involving pulmonary vasodilators proven efficacious for PAH have either failed or shown harm in patients with hypoxic PH. Further, there is a subset of hypoxic PH patients that develop severe “out-of-proportion” PH characterized by pulmonary vascular remodeling that is irreversible and similar to that in WHO Group 1 disease. In both mild to moderate as well as severe forms of the disease, inflammation appears to play an important role in the disease process. In this review, we highlight the mechanisms involved in both the initiation and perpetuation of inflammation in the vessel wall as well as the interplay between hypoxia, inflammation and their effects on resident pulmonary vascular cells and recruited immune and progenitor cells. We hypothesize that those patients with severe, “out of proportion” and irreversible hypoxic PH, experience an as yet undefined “second hit” whereby the mechanisms involved go beyond those related to hypoxia alone, leading to chronic non-resolving inflammation. We propose that important epigenetic changes, at least in mesenchymal cells, and possibly in perivascular macrophages, are paramount in “locking” cells into a pro-remodeling, pro-inflammatory, and pro-mitogenic phenotype. Furthermore, these fibroblast/macrophage-stromal cell interactions are necessary for the maintenance of chronic non-resolving inflammation and persistent pulmonary vascular remodeling. After nearly 20 years of vasodilator therapies that have reduced morbidity in patients with PAH, mortality remains unacceptably high, forcing the field to move toward treatments targeting the underlying pathogenesis of pulmonary vascular remodeling. Similarly, the persistent failure of pulmonary vasodilators in hypoxic PH urges us to shift away from recycling pulmonary vasodilator therapies proven to be beneficial in PAH to those that aim to disrupt and repair the basic pathologic inflammatory mechanisms responsible for the initiation and perpetuation of disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


