The Dyslipidemias
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality in the industrialized world, accounting for approximately one-third of all deaths in the United States. Coronary heart disease (CHD), a subcategory of CVD, kills nearly 1 in every 5 Americans. Each year, 1.5 million people in the United States suffer a first or recurrent myocardial infarction (MI), and nearly 325,000 people experience sudden cardiac death (SCD). Stroke remains the third leading cause of mortality with 800,000 strokes per year in the United States. Despite reductions in mean serum total cholesterol (TC) from 222 mg/dL in 1962 to 199 mg/dL in 2006, nearly half of US adults still have abnormally high levels. Given the enormous burden of CVD, the high prevalence of lipid disorders, and effective evidence-based treatment strategies, recognition of and management of lipid disorders is an essential component of both primary and secondary prevention of CVD. In this chapter, we have sought to provide a clinically relevant discussion of dyslipidemias and their effective treatment to reduce CVD morbidity and mortality. In addition to defining lipoproteins and lipid disorders, the clinical trials and observational studies that form the cornerstone for modern treatment guidelines are reviewed.
LIPIDS AND LIPOPROTEINS
Lipids are molecules with hydrocarbon skeletons, which play crucial roles in the storage, metabolism, and production of energy, the structure and behavior of cell membranes, and the transduction of signals both inside and between cells. Lipids are fat-soluble, or lipophilic, compounds, which are classified as either simple or complex. Simple lipids include free cholesterol (FC) and fatty acids (FAs), and complex lipids, which are combinations of simple lipids, include cholesteryl esters (CEs) and triglycerides (TG). The lipids with greatest pathologic significance appear to be CE, the predominant components of macrophage foam cell inclusions, and TG, which form the core of adipocyte inclusions. Packaged together with phospholipids and proteins known as apolipoproteins, lipids are transported between organs in the form of lipoproteins. Lipoproteins are classified according to their densities and electrophoretic mobilities and include, in decreasing density, high-density lipoproteins (HDL), low-density lipoproteins (LDL), intermediate-density lipo-proteins (IDL), very low-density lipoproteins (VLDL), and chylomicrons.
Lipid processing and transport can be envisioned as a bidirectional process. The first cycle begins with triglyceride-rich lipoproteins (TGRLs), namely, chylomicrons from the gut and VLDL from the liver (Fig. 53.1). These lipoproteins serve as substrates for lipoprotein lipase (LPL) and hepatic lipase (HL), two different enzymes bound to capillary endothelium. Free FAs are released for the use of skeletal muscle, adipose tissue, and other organs and FC and CE are delivered to distant tissues. Chylomicrons, VLDL, and their lipolysis products, such as chylomicron remnants, IDL, and LDL, bear apolipoprotein B (apoB) and apolipoprotein E (apoE) on their surfaces. ApoB has two isoforms: apoB100, which is present on lipoproteins secreted by the liver, and apoB48, which is present on gut-derived lipoproteins. LDL, IDL, and remnant particles can be taken up by hepatocytes via receptors that bind to apoB and apoE. Alternatively, they may migrate across the endothelium, undergo oxidation/modification, induce monocyte recruitment and foam cell formation, and initiate a cycle of inflammation, chemotaxis, and thrombotic activity leading to atherosclerosis and vascular events. IDL is the most atherogenic lipoprotein, but LDL (which carries 70% of plasma cholesterol) is the principle lipoprotein responsible for atherosclerosis. Cholesterol present in apoB-containing lipoproteins may be represented as non–high-density lipoprotein cholesterol (non-HDL-C). Small dense LDL (sdLDL) is defined as LDL with a low LDL-C to LDL-apoB ratio (<1.2) and represents a more atherogenic subclass of LDL.
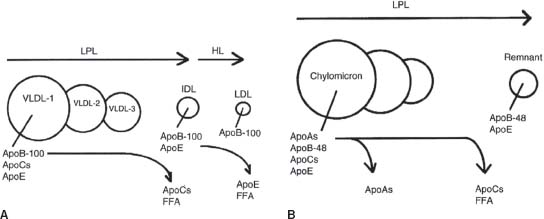
FIGURE 53.1 Remodeling of TGRLs. A: Stepwise lipolysis of VLDL subfractions 1, 2, and 3 by LPL. LPL catalyzes VLDL triglyceride hydrolysis with the concomitant transfer of the apoC proteins to HDL and the release of free fatty acids (FFA). Subsequently, HL catalyzes additional triglyceride hydrolysis that induces the transfer of apoE from IDL to HDL and the release of additional FFA. LDL contains apoB100 as its sole protein. B: Lipolysis of chylomicrons by LPL. Triglyceride hydrolysis is associated with the transfer of the apoC proteins to HDL and the release of FFA. The remnant contains apoB48, which is the major protein of chylomicrons, and apoE, which mediates the binding of remnants to hepatic receptors. (Reprinted from Betteridge DJ, Illingworth DR, Shepherd J. Lipoproteins in Health and Disease. 1st ed. London: Arnold, 1999:4, with permission.)
The other major transport cycle, sometimes termed reverse cholesterol transport (RCT), serves primarily to return cholesterol to the liver (Fig. 53.2). RCT involves HDL, the major structural protein of which is apolipoprotein A (apoA). Starting with a lipid-poor HDL known as pre-β HDL, FC is progressively added from macrophages, and then esterified by the enzyme lecithin cholesterol acyltransferase (LCAT). These mature HDL-cholesterol (HDL-C) particles can return to the liver by direct hepatic uptake of HDL, or CE can be transferred to apoB-containing lipoproteins, which then can be taken up by the liver. One clinically relevant reaction is catalyzed by the cholesteryl ester transfer protein (CETP), which transfers CE from HDL to LDL or VLDL in exchange for TG. Thus, CE and TG are rapidly equilibrated between HDL and the apoB-containing lipoproteins, a major reason why patients with hypertriglyceridemia tend to have low HDL-C. The subsequent action of HL on TG-rich HDL leads to the production of smaller, denser HDL in such individuals. Individuals with diminished or absent levels of serum CETP generally have elevated HDL-C and lower rates of atherosclerosis. Pharmacologic inhibitors of CETP have been shown to significantly elevate HDL-C, but initial studies showed harm, not benefit. However, other CETP drugs “in the pipeline” may yet become important tools for the clinician. In addition to its role in lipid transport, HDL may mediate other antiatherosclerotic effects by inhibiting LDL oxidation, reducing endothelial dysfunction, reducing chemotaxis of inflammatory cells into plaques, and inhibiting thrombosis.
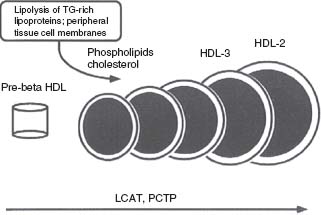
FIGURE 53.2 Remodeling of HDL by LCAT and phosphatidylcholine transfer protein (PCTP, also known as phospholipid transfer protein or PLTP). Small, premigrating HDL composed of apoA-I, cholesterol, and lecithin is a substrate for LCAT, which forms CEs within the core. Additional cholesterol and phospholipids from peripheral tissue cell membranes and lipolysis of TGRLs are added to the HDL. Multiple cycles of lipid transfer to HDL and LCAT activity eventually produce large, mature HDL, which is a major carrier of cholesterol to the liver. (Reprinted from Betteridge DJ, Illingworth DR, Shepherd J, eds. Lipoproteins in Health and Disease. 1st ed. London: Arnold, 1999:4, with permission.)
Lipoprotein (a), or Lp(a), is an LDL-like particle of hepatic origin. Unlike LDL, apoB100 on the surface of Lp(a) is bound to a protein called apolipoprotein (a), or apo(a), which has strong homologies to plasminogen. While apo(a) lacks enzymatic activity, Lp(a) can interfere with the binding of plasminogen to substrates such as fibrin, cell surfaces, and extracellular matrix, potentially promoting a prothrombotic state. Numerous retrospective and prospective studies have shown a clear association between high plasma Lp(a) levels and CHD, and such associations appear to be genetically mediated. At this time, though, we still lack evidence that pharmacologic reduction of Lp(a) levels can lower the incidence of coronary events.
DIAGNOSIS OF DYSLIPIDEMIAS
Clinical interest is focused on dyslipidemias in which a causal or proposed causal relationship exists between abnormal serum lipid levels and atherosclerosis. Appropriate treatment of a particular dyslipidemia requires accurately characterizing a patient’s lipid disorder. In order to classify a patient’s dyslipidemia and exclude secondary causes and determine treatment strategies, the clinical practitioner should investigate the following:
 Personal history: abnormal serum lipid levels; CHD; manifestations of cerebrovascular disease, including transient ischemic attack (TIA) or cerebrovascular accident (CVA); peripheral vascular disease (PVD), including limb claudication, aortic aneurysm, or carotid atherosclerosis; diabetes mellitus (DM); hypothyroidism; chronic renal insufficiency (CRI); nephrotic syndrome; hepatobiliary disease; pancreatitis; or pregnancy
Personal history: abnormal serum lipid levels; CHD; manifestations of cerebrovascular disease, including transient ischemic attack (TIA) or cerebrovascular accident (CVA); peripheral vascular disease (PVD), including limb claudication, aortic aneurysm, or carotid atherosclerosis; diabetes mellitus (DM); hypothyroidism; chronic renal insufficiency (CRI); nephrotic syndrome; hepatobiliary disease; pancreatitis; or pregnancy
 Medication history: especially of thiazide diuretics, β-blockers, oral contraceptives (OCPs), hormone replacement therapy (HRT), isotretinoin, glucocorticoids, or highly active antiretroviral therapy (HAART) for HIV
Medication history: especially of thiazide diuretics, β-blockers, oral contraceptives (OCPs), hormone replacement therapy (HRT), isotretinoin, glucocorticoids, or highly active antiretroviral therapy (HAART) for HIV
 Family history: dyslipidemias, CHD, TIA/CVA, PVD, sudden death, diabetes, hypertension, or central obesity. Suspicion of familial dyslipidemias should be followed up with lipid testing of family members.
Family history: dyslipidemias, CHD, TIA/CVA, PVD, sudden death, diabetes, hypertension, or central obesity. Suspicion of familial dyslipidemias should be followed up with lipid testing of family members.
 Lifestyle history: past and present tobacco use, excessive alcohol use (>40 g/d), sedentary lifestyle, or diets rich in saturated fats or carbohydrates
Lifestyle history: past and present tobacco use, excessive alcohol use (>40 g/d), sedentary lifestyle, or diets rich in saturated fats or carbohydrates
 Physical exam findings: body mass index (BMI), waist circumference, blood pressure, thyroid characteristics, xanthomas (cholesterol deposits of interdigital, tuberous, planar, or eruptive types), xanthelasmas (xanthomas of the palpebral fissures), arcus corneae, lipemia retinalis (pale-appearing retinal vessels), peripheral pulses, vascular bruits, or hepatosplenomegaly. See Figure 53.3 for representative examples of xanthomas
Physical exam findings: body mass index (BMI), waist circumference, blood pressure, thyroid characteristics, xanthomas (cholesterol deposits of interdigital, tuberous, planar, or eruptive types), xanthelasmas (xanthomas of the palpebral fissures), arcus corneae, lipemia retinalis (pale-appearing retinal vessels), peripheral pulses, vascular bruits, or hepatosplenomegaly. See Figure 53.3 for representative examples of xanthomas
 Laboratory studies: fasting serum lipid panel, fasting glucose, thyroid-stimulating hormone (TSH), creatinine, hepatic function panel, urinalysis, and a screen for microalbuminuria. Additional measurements such as lipoprotein subpopulation analysis, levels of serum Lp(a), apoB, and high-sensitivity CRP (hsCRP) may help refine risk assessment, particularly in an intermediate-risk population or to help tailor intensity of therapies, but would not be recommended as routine tests for all individuals.
Laboratory studies: fasting serum lipid panel, fasting glucose, thyroid-stimulating hormone (TSH), creatinine, hepatic function panel, urinalysis, and a screen for microalbuminuria. Additional measurements such as lipoprotein subpopulation analysis, levels of serum Lp(a), apoB, and high-sensitivity CRP (hsCRP) may help refine risk assessment, particularly in an intermediate-risk population or to help tailor intensity of therapies, but would not be recommended as routine tests for all individuals.
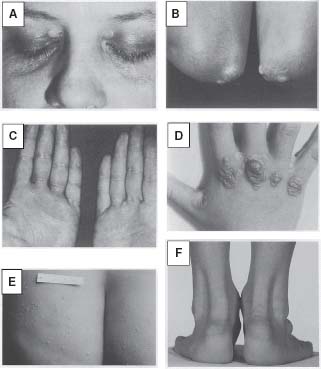
FIGURE 53.3 Representative xanthomas are illustrated: A: Xanthelasmas (xanthomas of the palpebral fissures). B: Tuberous xanthomas of the elbows. C: Palmar xanthomas. D: Interdigital xanthomas. E: Tuberoeruptive xanthomas of the buttocks. F: Tendon xanthomas of the Achilles tendon. (Reprinted from Fuster V, Ross R, Topol EJ, eds. Atherosclerosis and Coronary Artery Disease. 1st ed. Philadelphia: Lippincott-Raven; 1996, with permission.)
CLASSIFICATION OF DYSLIPIDEMIAS
In 1967, Fredrickson et al.1 proposed a system to diagnose and classify lipid disorders based upon the specific lipoprotein or lipoproteins elevated in the patient’s serum. While the Fredrickson classification system was useful shorthand for describing hyperlipidemias, it had two major shortcomings: its phenotypes did not provide or even imply etiology, and HDL was not included. In a more clinically relevant schema, lipid disorders can also be grouped into two broad categories. Primary dyslipidemias result from genetic variability in one or more loci controlling the expression of proteins involved in lipoprotein synthesis, metabolism, or clearance. Secondary dyslipidemias are the consequence of a separate pathologic process.
Primary Dyslipidemias
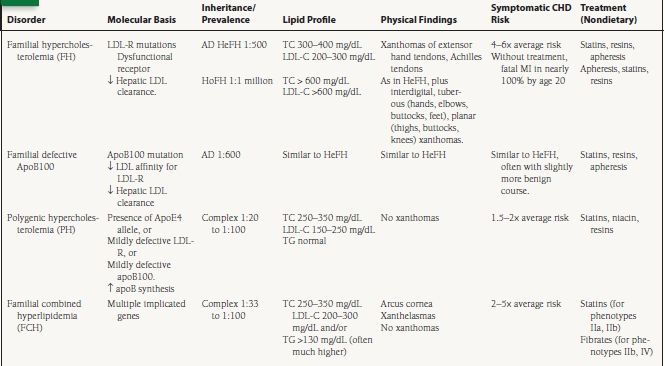
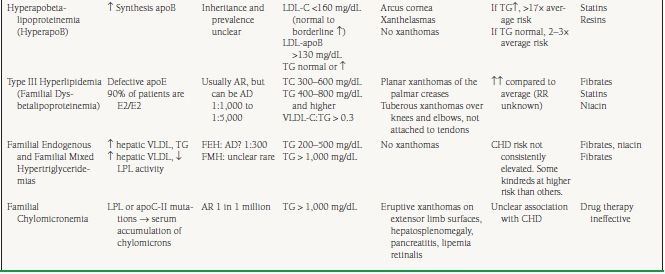
The primary dyslipidemias of non-HDL-C have been summarized in Table 53.1. Familial hypercholesterolemia (FH) is a common autosomal dominant disorder resulting from mutations in the LDL receptor (LDL-R), or apoB receptor, leading to impaired hepatic clearance of LDL from the circulation. Heterozygous FH (HeFH) occurs in 1 in 500 persons and is associated with serum LDL-C two to three times the average and a four- to sixfold increased risk for premature CHD. Without treatment, the average age for development of symptomatic CHD is 45 years in men, and 55 years in women. By age 39, 90% of FH heterozygotes exhibit detectable xanthomas on the extensor tendons of the hands, or on the Achilles tendons (Fig. 53.3). Several diagnostic criteria for FH exist, with that of the 15-year Simon Broome Register Group being the most commonly used.
TABLE
53.1 Primary Dyslipidemias of Non-HDL-C
AD, autosomal dominant; AR, autosomal recessive; HeFH, heterozygous FH; HoFH, homozygous FH; RR, relative risk.
(a) TC > 290 mg/dL in adults or TC > 260 mg/dL in children under 16
OR LDL-C > 190 mg/dL in adults or >155 mg/dL in children
PLUS
(b) Tendon xanthomas in the patient, or first- or second-degree relative
(c) OR DNA-based evidence of LDL-R mutation or familial defective apoB100
Possible FH is defined as (a) above plus one of (d) or (e):
(d) MI before age 50 in second-degree relative, or before 60 in first degree
(e) Elevated cholesterol in first-degree relative, or >290 mg/dL in second degree
Homozygous FH (HoFH) occurs in one in one million individuals. Patients with HoFH do not express any functional LDL-Rs, and consequently exhibit a more severe phenotype. TC levels are generally >600 mg/dL, with LDL-C levels six- to eightfold higher than average. Without treatment, death from MI occurs in the first or second decades of life. In addition to the xanthomas observed in heterozygotes, FH homozygotes are commonly affected by interdigital xanthomas, tuberous xanthomas on the hands, elbows, buttocks, and feet, and planar xanthomas on the posterior thighs, buttocks, and knees.
Familial Defective Apolipoprotein B100 is associated with impaired LDL clearance due to reduced affinity of LDL for the LDL-R. The most common cause of this autosomal dominant condition is a single base mutation in the apoB100 gene. Although the prevalence of familial defective apoB100 is unclear, and varies by ethnic background, it may be as common as 1 in 600. The lipoprotein concentrations, clinical features, and treatment are similar to FH heterozygotes.
Polygenic hypercholesterolemia (PH) is the most common cause of an isolated elevation in TC or LDL-C, with prevalence in the United States estimated at between 1 in 20 and 1 in 100. TG levels are generally normal. Alterations in the function or expression of several key proteins involved in LDL metabolism have been associated with PH, including mildly defective LDL-R and apoB100, increased synthesis of apoB, and the presence of the apoE4 allele, which has a higher affinity for the LDL-R than the other apoE isoforms leading to downregulation of LDL-R synthesis and a secondary increase in serum LDL-C. Xanthomas are very rare or absent in patients with PH.
Familial combined hyperlipidemia (FCH) is the most common primary dyslipidemia in which multiple lipoprotein phenotypes exist, with a population prevalence of 1% to 3%. The three observed patterns of elevated VLDL, elevated LDL, or both can be seen within a family or within a single patient over time. Transmission is complex, with multiple genes likely to be involved. It is associated with an estimated two- to fivefold increased risk for CHD, accounting for one-third to one-half of familial CHD, and up to 20% of all premature CHD. Traditionally, diagnostic criteria include:
1. TC and/or TG levels >90th percentile for age- and sex-matched controls. TC of 250 to 350 mg/dL and TG > 130 mg/dL (often higher, especially in diabetics).
2. At least one first-degree relative with elevated VLDL-C, LDL-C, or both
3. Strong family histories of hyperlipidemia and premature CHD
Other major characteristics include elevated apoB (>120 mg/dL), a preponderance of sdLDL, and low HDL-C. FCH is usually diagnosed after age 20, and the patients are often hypertensive, overweight, insulin resistant, or diabetic. Arcus cornea and xanthelasmas are commonly seen, but tendon xanthomas are unusual. Hence, the absence of tendon xanthomas in a hypercholesterolemic patient is a useful feature to differentiate FCH from FH.
Other less common dyslipidemias of apoB metabolism are frequently characterized by elevations in TG, VLDL or in LDL-apoB concentrations. These include hyperapobetalipo-proteinemia (hyperapoB), type III hyperlipidemia (also known as familial dysbetalipoproteinemia or remnant disease), familial endogenous hypertriglyceridemia (FEH), familial mixed hypertriglyceridemia (FMH), and familial chylomicronemia and are outlined in Table 53.1.
There exist a heterogeneous group of rare familial disorders of HDL-C, including Tangier Disease, Familial LCAT Deficiency, and Partial LCAT Deficiency or Fish-Eye Disease which have not been consistently associated with premature CHD and are beyond the scope of this chapter. ApoAIMilano is a rare genetic variant of the apoAI protein resulting in low HDL-C levels. However, individuals bearing the mutation display longevity and an exceptionally low risk of CHD. In a controlled trial of patients with a history of acute coronary syndrome (ACS), infusions of purified ApoAIMilano induced plaque regression, as assessed by intravascular ultrasound (IVUS). At present, such therapy is not available for clinical use but these studies suggest a future role for HDL-C modification to reduce cardiovascular risk.
Secondary Dyslipidemias
Evaluation of a dyslipidemia would not be complete without a thorough search for secondary and contributing causes. A careful history and physical, accompanied by selected laboratory studies, including a fasting lipid panel, will frequently reveal the etiology of a patient’s dyslipidemia. Breakdown by the abnormal lipid may simplify diagnosis of the dyslipidemia’s cause:
 Elevated TC and LDL-C: diet rich in saturated fat, drugs (oral contraceptives or OCPs, HRT, HAART), hypothyroidism, nephrotic syndrome, chronic liver disease, and chronic biliary tract disease (classically, primary biliary cirrhosis)
Elevated TC and LDL-C: diet rich in saturated fat, drugs (oral contraceptives or OCPs, HRT, HAART), hypothyroidism, nephrotic syndrome, chronic liver disease, and chronic biliary tract disease (classically, primary biliary cirrhosis)
 Elevated TG: diet rich in carbohydrates, drugs (β blockers, thiazide diuretics, isotretinoin, glucocorticoids, HAART, OCP, HRT), excessive alcohol consumption, obesity, DM, hypothyroidism, CRI, chronic pancreatitis, and pregnancy
Elevated TG: diet rich in carbohydrates, drugs (β blockers, thiazide diuretics, isotretinoin, glucocorticoids, HAART, OCP, HRT), excessive alcohol consumption, obesity, DM, hypothyroidism, CRI, chronic pancreatitis, and pregnancy
 Low HDL-C: very high carbohydrate, very low-fat diet, hypertriglyceridemia, obesity, sedentary lifestyle, smoking, DM
Low HDL-C: very high carbohydrate, very low-fat diet, hypertriglyceridemia, obesity, sedentary lifestyle, smoking, DM
Often, these dyslipidemias can be at least partially controlled by institution of lifestyle changes (including dietary improvement, weight loss, increased exercise, and smoking cessation), withdrawal and replacement on an implicated medication, or recognition and treatment of an underlying disorder. A typical example is the hyperlipidemic patient who is resistant to lipid-lowering therapy, but later found to be hypothyroid. Treatment with levothyroxine then corrects the secondary dyslipidemia.
METABOLIC SYNDROME
Due to the growing recognition of its prevalence and the association of this syndrome with a particular dyslipidemic pattern, the metabolic syndrome (MetS) is reviewed here. During the late 1980s, Reaven observed that several CHD risk factors (namely dyslipidemia, hypertension, and hyperglycemia) frequently cluster together.2 Originally referred to as Syndrome X or the insulin-resistance syndrome, these descriptors have been supplanted by the more general term metabolic syndrome. The National Cholesterol Education Program’s Adult Treatment Panel III report (ATP III, discussed later) identified six components of the MetS which relate to CHD: abdominal obesity, atherogenic dyslipidemia (high TG, low HDL-C, and sdLDL), hypertension (HTN), insulin resistance, a prothrombotic state, and a proinflammatory state.3 Given the ongoing “epidemic of obesity,” MetS is common and becoming more so, with approximately one-third of American adults fulfilling ATP III and global criteria,4 as listed in Table 53.2. Observational data have shown that such persons have a three to fivefold risk of CHD mortality, compared to those without MetS. A major health danger to persons with MetS appears to be a vastly increased risk of developing diabetes. MetS was highly predictive of new-onset DM in both men and women of the Framingham offspring cohort, as nearly half of the population-attributable risk of type 2 diabetes (DM2) could be explained by the presence of ATPIII-defined MetS.5 When overt DM develops, CHD risk increases sharply.
TABLE
53.2 ATP III Clinical Identification of the MetS
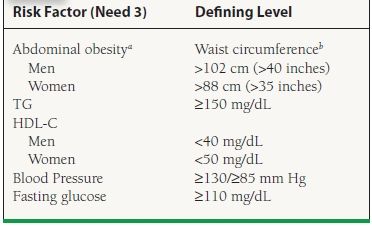
aOverweight and obesity are associated with insulin resistance and the MetS. However, the presence of abdominal obesity is more highly correlated with the metabolic risk factors than is an elevated BMI. Therefore, the simple measure of waist circumference is recommended to identify the body weight component of the MetS.
bSome male patients can develop multiple metabolic risk factors when the waist circumference is only marginally increased, for example, 37 to 39 inches (94 to 102 cm). Such patients may have a strong genetic contribution to insulin resistance. They should benefit from changes in life habits, similar to men with categorical increases in waist circumference. Adapted from the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on the Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (ATP III), Final Report, 2002. A publication of the National Heart, Lung, and Blood Institute (NHLBI), a division of the National Institutes of Health (NIH), and the U.S. Department of Health and Human Services.
TREATMENT STRATEGIES FOR DYSLIPIDEMIAS: THERAPEUTIC LIFESTYLE CHANGES
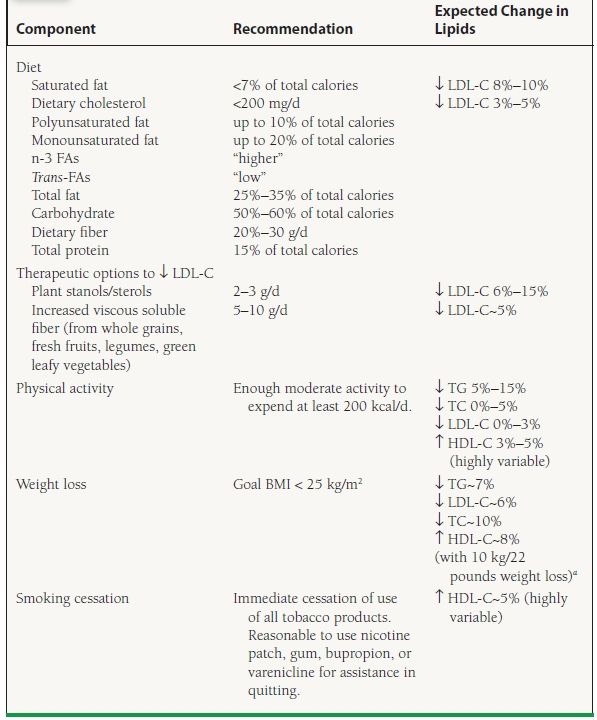
All dyslipidemic patients should be urged to adopt therapeutic lifestyle changes (TLC), consisting of increased physical activity, ideal weight maintenance (often necessitating weight loss), smoking cessation, and the pursuance of a low-saturated fat, low-cholesterol diet rich in fruits, vegetables, grains, and fiber (Table 53.3). In highly motivated individuals, TLC can result in an LDL-C reduction of nearly 30%, and should form the basis of all preventive treatment. Alcohol avoidance, smoking cessation, physical activity, and diet are essential in the management of dyslipidemias characterized by very high TG.
TABLE
53.3 Therapeutic Lifestyle Changes
Adapted from the ATP III Final Report (2002).
aDattilo AM, Kris-Etherton PM. Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am J Clin Nutr. 1992;56:320–328.
Most primary dyslipidemias other than HoFH, as well as the dyslipidemia associated with MetS, are very sensitive to changes in diet and adiposity, and thus should always be treated with a TLC diet and weight management, with a goal BMI <25 kg/m2. Lifestyle changes are first-line therapy for the MetS, with obesity the primary target of intervention.6 The most critical goal of such modifications is to decrease the incidence of new-onset DM, which confers a similar risk of CHD events as known CHD. It may be challenging to successfully implement such changes, but every effort should be made to stress the clear health benefits of dietary modification, weight loss, and physical activity.
Whether TLC are sufficient alone, or should be accompanied by drug therapy, is determined by the dyslipidemic patient’s CHD risk status, initial lipid levels, and treatment targets (to be discussed in later sections). Other nonpharmacologic approaches such as dietary intake of plant stanols/sterols (in certain labeled margarines and juices and pill supplements) and increased viscous soluble fiber (in oats, barley, psyllium, pectin-rich fruits, and legumes) may also aid in LDL-C reduction. High-dose omega-3 fish oil supplementation may aid in TG lowering. Consultation of a registered dietician or qualified nutritionist may be useful. In high-risk individuals pharmacologic therapy should be introduced simultaneously with TLC. In others, if goal LDL-C has not been reached after 3 months, pharmacologic therapy should be considered.
TREATMENT STRATEGIES FOR DYSLIPIDEMIAS: PHARMACOLOGIC THERAPY
Pharmacologic therapy should be initiated in patients whose lipids are inadequately lowered with TLC alone, those with lipids too high to be reasonably reduced with TLC alone, or those with CHD risk high enough to mandate drug initiation simultaneously with TLC. There are six major classes of drugs which can be used to regulate a patient’s lipid profile, including (a) HMG-CoA reductase inhibitors, or “statins,” (b) bile acid sequestrants or “resins,” (c) fibric acid derivatives or fibrates, (d) nicotinic acid or niacin, (e) cholesterol absorption inhibitors of which ezetimibe is the only clinically available member, and (f) Omega-3 FAs of which Lovaza is the only available prescription formulation. Table 53.4 includes a description of these agents, their mechanisms of action, therapeutic indications and contraindications, effects on lipid levels, and adverse effects. The choice of a specific drug or combination of drugs is dependent on an understanding of each medication’s mechanism of action, as well as the individual patient’s lipid profile, cardiovascular risk, treatment goals, and contraindications. Medication choice should also be influenced by clinical outcome trials demonstrating reduction of cardiovascular events with specific treatments. Specific treatment guidelines are reviewed in a later section. Fasting lipid levels should be checked 6 weeks after drug initiation. Once the treatment goal has been achieved, fasting lipids should be redrawn every 4 to 6 months. Importantly, TLC should always be used concomitantly with drug therapy of dyslipidemias. Referral to a lipid specialist should be considered for complex combined lipid disorders or nonresponders.
TABLE
53.4 Lipid-Regulating Drugs
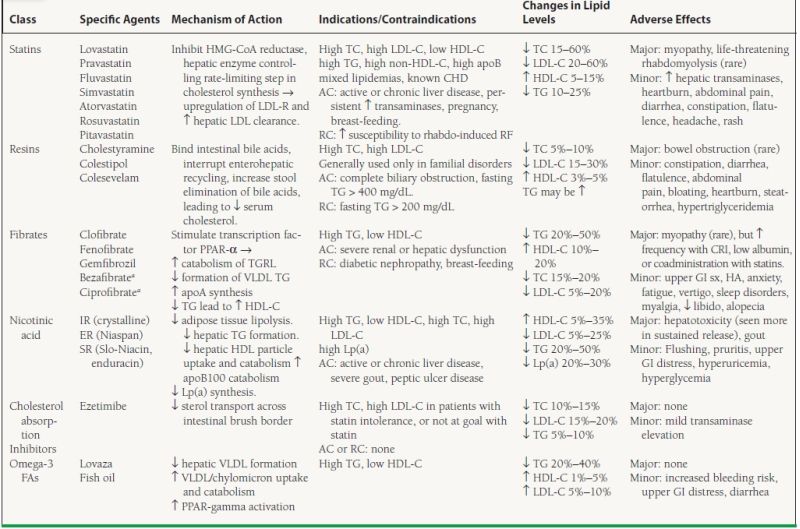
anot available in the U.S.; AC, absolute contraindication; RC, relative contraindication; CRI, chronic renal insufficiency; HA, headache; IR, immediate release; ER, extended release; SR, sustained release
Of the primary dyslipidemias, treatment of HeFH includes dietary approaches and aggressive pharmacologic therapy. Since the hepatocytes of these individuals still express the LDL-R, albeit at a lower concentration than normal, they are able to upregulate its level and thus increase clearance of LDL. Multiple studies involving adults with HeFH have shown high-dose statin therapy to be safe, well tolerated, and effective at reducing LDL-C levels, CHD morbidity/mortality. Children and adolescents with HeFH are also effectively treated with statins. Despite concerns over possible interference with hormonal pathways, the growth parameters and sexual maturation in statin-treated children were similar to those given placebo.7 Often, combination drug therapy of a statin with ezetimibe, resins, and/or niacin is necessary to adequately lower LDL-C levels. As for HoFH, dietary changes are not effective at reducing LDL-C. Given the complete absence of functional LDL-Rs, one would not predict that FH homozygotes could be treated effectively with statins. However, multiple small studies have shown that statins reduce LDL-C by 15% to 35% in such persons, probably via decreased hepatic synthesis of VLDL and LDL.
Drug treatment of persons with other primary dyslipidemias is determined largely by their individual lipid profiles (see Table 53.1). In general, elevated LDL-C should be treated with a statin unless contraindicated or not tolerated. Disorders of elevated TG are usually evaluated with a search for secondary causes, and treated with dietary modification, physical activity, and fibrates, niacin or omega-3 FAs. The dyslipidemia associated with MetS should always be initially treated with lifestyle approaches, especially weight loss. However, failure to achieve full reversal of the MetS characteristics may necessitate pharmacologic therapy. Subgroup analyses of statin trials have shown that statins reduce risk for CVD events in MetS patients. A post hoc analysis of recent fibrate trials strongly suggests that they reduce CVD endpoints in patients with atherogenic dyslipidemia and MetS.
In some cases, drugs from multiple classes used in combination may be required for adequate LDL-C lowering and/or treatment of combined dyslipidemias. Studies have supported acceptable tolerance and improvement of the lipid profile with combination therapy in certain subgroups. Although lipoprotein improvements have been even more dramatic on combined statin–fibrate, statin–niacin, or stain-resin/ezetimibe therapy, no CVD endpoint data from large, controlled trials are available. Smaller trials and those employing surrogate vascular endpoints suggest clinical benefit of combination therapy. It is important to be familiar with the adverse effects of lipid-regulating drugs (Table 53.4). Such adverse effects may occur more frequently with combination therapy, including rhabdomyolysis (more common with statin + fibrate) and hepatic injury (statin + niacin, statin + ezetimibe, statin + fibrate). Obese patients undergoing weight-loss (bariatric) surgery are observed to exhibit a dramatic decline in clinical features of MetS. Their total and LDL-C tend to be significantly lower. Most impressively, a systematic review has also demonstrated that most diabetic patients undergoing such surgery experience total postoperative reversal of their diabetes diagnosis.
APHERESIS THERAPY
The mainstay of therapy for FH homozygotes is extracorporeal, namely LDL apheresis. On average, LDL-C levels immediately after the procedure are decreased 50% to 80%. Since these values rebound fairly quickly, the process is performed every 2 weeks to keep intrapheresis LDL-C ≤120 mg/dL. FDA indications since 1996 include: HoFH; HeFH in the absence of CHD when LDL-C ≥ 300 mg/dL despite maximal pharmacologic and dietary therapy; and HeFH in the presence of CHD when LDL-C ≥ 200 mg/dL despite maximal pharmacologic and dietary therapy. The benefits of apheresis to FH homozygotes, in terms of stabilization or regression of atherosclerotic lesions, and improvement in symptoms, have been clearly demonstrated. As for FH heterozygotes, combined apheresis and statin therapy have been shown to substantially reduce risk of coronary events, and to improve angiographic outcome.
LIPID-REGULATING TRIALS FOR THE PREVENTION OF CHD
The rationale for aggressive management of lipid disorders for the purpose of reducing cardiovascular events is based on a large body of research spanning the past decade. A complete review of the research establishing an association between dyslipidemia and CHD is beyond the scope of this chapter, but it is important to highlight the observations and trials supporting recent cholesterol treatment recommendations. As early as the 1930s, associations were observed between cholesterol levels and atherosclerotic disease. Multiple, large observational and epidemiologic studies helped to form the basis of the cholesterol hypothesis, which posited that elevated serum cholesterol plays a causative role in the development of CHD, and that cholesterol reduction will reduce CHD risk.8 These studies included the Framingham Heart Study, the Seven Country Study, the Münster Heart Study (PROCAM), the Multiple Risk Factor Intervention Trial (MRFIT), and the Lipid Research Clinics (LRC) Prevalence Study. Data from these studies revealed a strong, graded, linear relationship between TC levels and risk of CHD, with an approximate 20% to 30% increase in CHD risk for each 10% increase in serum TC. An inverse correlation was observed for HDL-C, with every 1 mg/dL (0.026 mM) increase in HDL-C correlating with a 2% decrease in CHD risk for men, and a 3% decrease for women.9 Hypertriglyceridemia was long suspected to be an independent CHD risk factor, but this was difficult to prove given the tight inverse correlation between levels of TG and HDL-C. A meta-analysis of 17 population-based studies showed that each 1 mmol/L (88.5 mg/dL) increase in serum TG significantly increased the risk of a CHD event, by 32% in men, and 76% in women. After multivariate analysis, including adjustment for HDL-C levels, a 1 mmol/L increase in TG continued to confer a significantly increased CHD risk, by 14% in men, and 37% in women.
Once these relationships had been identified, clinical trials were designed to test the hypothesis that lipid-lowering therapy would slow or reverse the atherosclerotic process and decrease the incidence of CVD events. Despite the diversity in entry criteria and treatments, initial animal and human intervention studies demonstrated plaque regression or improvements in angiographic outcomes associated with improved lipid profiles. Although some of these trials did demonstrate decreased coronary events, the primary endpoints were typically surrogate outcomes and the trials were not designed or powered to examine clinical outcomes. Subsequent intervention trials targeted “hard” clinical endpoints, including death and nonfatal MI. In primary prevention trials, individuals without known CVD underwent cholesterol reduction, with the goal of preventing a first CHD event. In secondary prevention trials, subjects with known CVD were treated to lower cholesterol, in an effort to prevent repeat events.
EARLY TRIALS FOR PRIMARY AND SECONDARY CHD PREVENTION
A number of CVD endpoint trials, reviewed in detail in Table 53.5, were performed in the prestatin era, utilizing diet, bile acid sequestrants, fibrates or niacin. These trials, in predominantly male populations both with and without CHD, using various treatments or combination of treatments, provided strong support for the cholesterol hypothesis, giving rise to the rule of thumb that a 1% lowering of TC decreases the incidence of CHD events by 2% to 3%. In several of these studies, noncardiovascular death was increased in the group treated with lipid-lowering therapy, raising concern over the safety of such treatment. However, a causal link was never established between lipid-lowering drugs and increased mortality. The early trials set the stage for the large statin trials of the 1990s.
TABLE
53.5 Early Lipid-Regulating Trials
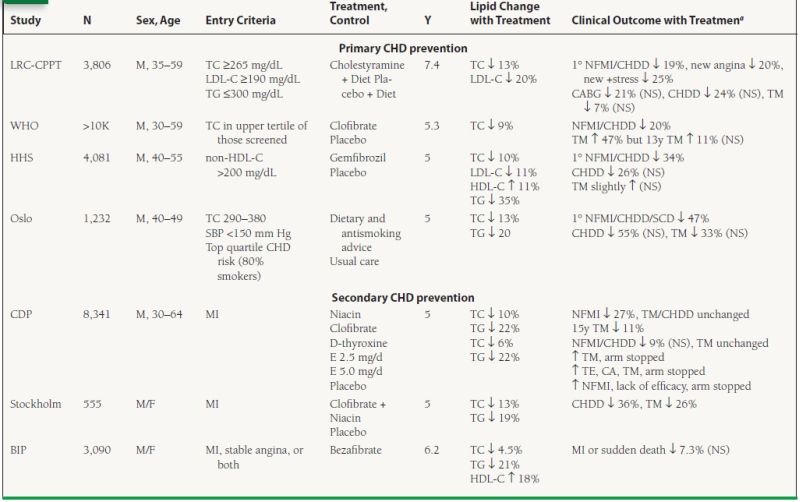
N, number of patients in trial; Y, years of trial duration; aall outcomes significant except as noted; 1°, primary endpoint; NFMI, nonfatal MI; CHDD, CHD death; SCD, sudden cardiac death; +stress, positive stress test; TM, total mortality; E, estrogen; NS, nonsignificant p-value.



