Hypoxic signaling
HIF-1α/HIF-2α
TGF-β
ERK1/2
Increased Ca++
Mitochondrial dysfunction
Mitochondrial hyperpolarization
Suppressed mitophagy
Increased mROS generation
HIF-1α and aerobic glycolysis
Quasi malignancy
Clonal endothelial cell proliferation
Microsatellite instability
Somatic chromosomal abnormalities
Dysregulated energetic metabolism
Inflammation
Perivascular T-cell and macrophages
Activated TGF-β signaling
Increased IL-6, IL4, IL-13
CCL5/RANTES
Pathological Characteristics of Intima, Media and Adventitial Remodeling
Vascular remodeling in PAH is characterized by smooth muscle cell proliferation, hypertrophy of the medial layer, arteriolar muscularization and endothelial cell proliferation. However, we will review the overall pathological changes in rodent and human vasculature that characteristically defines PAH. The pathological diagnosis of pulmonary vascular remodeling has benefited from the progressive use of cell-specific immunohistochemistry to better define the structure and cellular composition of the pulmonary vascular lesions through pathological interpretation of PH. Remodeling in pulmonary artery (PA) e.g., intima, media and adventitia is probably the major contributor to reduced cardiac output (CO) and enhanced pulmonary vascular resistance (PVR). Therefore, reduction of PA luminal area is probably the critical factor in the increase in PVR in PAH and other forms of PH [3].
In recent years, the Pulmonary Hypertension Breakthrough Initiative (PHBI) allowed us the unique opportunity to determine the pattern of pulmonary vascular remodeling in a large set of lung tissues with PAH [2]. There are numerous potential predictions regarding how pulmonary artery remodeling might be present in patients with the disease. Previously, we already have provided data regarding the relationship between the numbers of profiles of pulmonary vascular lesions and parameters of hemodynamics in PAH [4]. However, based on physiological coupling between the heart and lung during rest and exercise, it has been predicted that overall surface area could be required to decrease by 80 % by the time PH develops [2]; the specific contribution of pulmonary vascular remodeling vis-a-vis vasoconstriction is unclear. However, the PHBI cohort provided us with several key findings and advanced our understanding of the spectrum of the pathology of the disease.
Intima remodeling involves proliferation of endothelial cells (forming the plexiform lesions), accumulation of myofibroblasts (intima obliteration or fibrosis) and accumulation of extracellular matrix including collagen and mucopolysaccharides [5]. The intima remodeling, irrespective of specific proportions of each of these individual components, leads to almost complete obliteration of the vascular lumen, forming intima lesions, the most characteristic morphological finding in severe PH. These lesions are associated with congenital heart malformations and located at branching points, often as isolated lesions [6, 7]. Although plexiform lesions are seen predominantly in idiopathic PAH (IPAH), their precursor and importance as a trigger of severe PAH remains unclear. We speculate that endothelial cell proliferation and fibrosis in luminal area could be due to environmental insult as a key process that might precede occlusive intima lesions. The later consisted of eccentric intima thickening, fibrotic, plexiform, concentric, and dilation/angiomatoid lesions and represent a process of misguided angiogenesis based on the findings of expression of vascular endothelial growth factor (VEGF), its receptors 1 (flt) and 2 (kdr), and hypoxia inducible factor (HIF)-1α and β [8]. Focal eccentric lesions and thickening can be detected in normal lungs, but these lesions are more widespread and impinge to a larger extent on the vascular lumen in PH. However, variable degrees of eccentric thickening have been reported in cigarette smokers lungs associated with pulmonary endothelial cell dysfunction, with or without evidence of PH [9].
The potential role for early endothelial cell apoptosis in the pathogenesis of uncontrolled proliferation of pulmonary endothelial cells was first documented in the rat model of severe pulmonary hypertension caused by the combination of VEGF receptor blockade with SU5416 and chronic hypoxia [10]. Moreover, the role of endothelial cell apoptosis in the pathogenesis of PH was also extended to the monocrotaline model [11]. Initial endothelial cell apoptosis might favor the emergence of apoptosis-resistant endothelial cells, with potential for uncontrolled proliferation [10, 12]. Further, dysfunctional ECs can either release factors that stimulate smooth cell (SMC) proliferation or fail to produce agents that usually suppress proliferation of SMCs in response to growth factors, such as apelin [13, 14].
A prominent feature of vascular remodeling is medial thickening where medial smooth muscle cells elaborate extracellular matrix proteins, alters lumen size by contraction and relaxation [15]. The medial smooth muscle cell layer represents approximately 10–15 % of the outside diameter of normal muscularized pulmonary arteries, while it approaches 30–60 % of the outside diameter in vessels of IPAH lungs [16–19]. More precisely, we mentioned that medial smooth muscle cell hypertrophy is a characteristic pathological feature of PH that involves muscularized arteries (ranging between 70 and 500 μm in diameter), and precapillary vessels (below 70 μm in diameter) [5]. Although careful morphometric assessments of medial remodeling are still lacking non-IPAH PH, it is apparent that medial thickening occurs in mild/moderate or severe PH and in cases of normal individuals exposed to cigarette smoke with no evidence of PH [9]. Although media remodeling was correlated with hemodynamic parameters, three of the four quartiles of media remodeling in PAH were equally shared with control lungs, however, only the fourth quartile with more extensive media thickness fell well above the thickness seen in control lungs. Of note, intima lesions appeared more severe than intima remodeling seen in control lungs, the combination of intima and media remodeling had the most significant correlation with pulmonary artery pressures. Nevertheless, medial thickening probably plays an important role in the pathogenesis of PH, hitherto it is difficult to relate the morphological identification of medial remodeling to specific levels of pulmonary artery pressures, degrees of severity of PAH, or potential for response to vasodilators [5].
The vascular adventitia compartment acts as a pathobiological “processing center” for the retrieval, integration, storage and release of key regulators of vascular wall function in health and disease and thus, considered as the principal “injury-sensing site” of the pulmonary vessel wall [20]. The adventitia is mostly composed of fibroblasts, it also contains local and circulating progenitor cells that differentiate into smooth muscle and endothelial cells and contributes to pathophysiological changes in vascular structure [21, 22]. The normal adventitia represents approximately 15 % of the external diameter of pulmonary arteries larger than 50 μm in diameter. In IPAH arteries, the adventitial thickness increases to 28 % of artery diameter, predominantly due to collagen deposition [16]. Whether the adventitia is thickened or presents with a heterogeneous stromal cell population in other forms of PH remains unclear. Adventitia remodeling failed to correlate significantly with hemodynamics in our cohort of PAH lungs despite an earlier extensive study of remodeling in IPAH [16]. This discrepancy can be explained by methodological differences and the difficulty in precisely defining adventitia boundaries. Our data do not detract from the growing importance of the adventitia niche in coordinating media remodeling and perivascular inflammation, a key process in PH.
Inflammatory Pathology and Pathobiology of PAH
The pathophysiological mechanisms of pulmonary hypertension are not fully understood. Although the exact pathophysiology remains unknown, there is increasing evidence to suggest an important role for inflammation to the development of pulmonary hypertension in particular in PAH. There is a high correlation and statistically significant association between perivascular inflammation and pulmonary artery pressures and pulmonary vascular remodeling [4]. While inflammation in PAH has been relatively well described [23], whether it is cause or consequence in the pathogenesis of this disease remains unexplored. Recent studies have pointed Inflammation as a cause of PH as it precedes to altered immune processes leading to vascular remodeling that underlie the development of pulmonary arterial hypertension [24, 25]. Inflammation has been defined as a complex series of interactions among immunological soluble factors and cells that can arise in response to environmental insult to cardiopulmonary system [26].
Several animal models like monocrotaline (MCT), chronic hypoxia, and increased pulmonary blood flow have been studied to investigate pathological contribution of inflammation in the pathophysiology of PH. These animal models along with the mouse model of BMPR II gene deletion [27], the vasoactive intestinal polypeptide deletion model [28], the simian immunodeficiency virus macaque model (a model of HIV-PAH) [29], the mouse model of schistosomiasis-induced PAH [30] and the vascular endothelial growth factor (VEGF) receptor-2 blockade model using SU5416 revealed perivascular inflammatory cell infiltrates around remodeled vessels [10]. The common inflammatory cells involved consist mainly of bone-marrow-derived macrophages [31] immature dendritic cells (DCs) [32] and a minority of lymphocytes. Elevated serum and pulmonary cytokine and chemokine levels precede the development of pulmonary vascular remodeling.
Histopathology and Inflammatory Chemokines and Cytokines in PAH
Evidence from clinical and experimental studies suggests that inflammation is a key in progression of vascular remodeling in PAH. Lung biopsies from patients with PAH including animal models of PH showed by varying degrees of perivascular inflammatory infiltrates, comprising T- and B-lymphocytes, macrophages, dendritic cells, and mast cells compared with control vessels [33, 34]. Recently, correlations of the degree of perivascular inflammation score with intima plus media and adventitia thickness, respectively, and with mean pulmonary arterial pressure supports a role for perivascular inflammation in the processes of pulmonary vascular remodeling [3]. The increased prevalence of PAH in patients with various inflammatory diseases further indicates an important role for the inflammatory and immunological process in the pathogenesis of the disease [35, 36].
Consistent with an exaggerated acquired immunologic response in IPAH, T regulatory cells were decreased around remodeled pulmonary arteries. Overall, these findings support the concept of impaired immunity in both IPAH and APAH associated with collagen vascular disease; there is compelling evidence of or frank autoimmunity in the latter, and possibly in IPAH as well. The concept of an autoimmune [37] and inflammatory component to PH is supported by the presence of autoantibodies in patients with IPAH [38], the immunologic basis of collagen vascular diseases [39], and the presence of markers of inflammation systemically in patients with IPAH [23, 40]. Recent pathological and pathobiological assessment on human and animal models provide evidence that both pulmonary vascular cells and inflammatory cells are important local sources of chemokines and cytokines that can lead to pulmonary vascular remodeling in PAH [41, 42]. These include interleukin (IL)-1β, IL-6, IL-4, IL-13, TGF-β, monocyte chemoattractant protein-1, fractalkine, CCL5/RANTES, and tumor necrosis factor (TNF)-α.
Chemokines are small proteins, with a molecular weight of around 8–10 kilodalton (kDa), which act in cell signaling and/or as cytokines. Chemokines, like CCL2/MCP-1, CCL5/RANTES and CX3CL1/Fractalkine, play a significant role in the pathogenesis of PH. For instance, MCP-1 (CCL2) is produced by vascular cells that stimulate monocytes/macrophage activation and migration through chemokine (C-C motif) receptor mediated response; plasma and lung tissue of patients with IPAH showed elevated levels of MCP-1 [43]. Furthermore, pulmonary artery smooth muscle cells (PASMC) and endothelial cells (EC) from patients with IPAH overexpressed MCP-1 and exhibited exaggerated migratory and proliferative responses by increased levels of the chemokine (C-C motif) receptor. Further, MCP-1-blocking antibodies blocked the migratory and proliferative response [43]. Likewise, RANTES (or CCL5) mediated the trafficking and homing of T-lymphocytes, monocytes monocytes, basophils, eosinophils, and natural killer cells through different chemokine receptors [44]. Moreover, Fractalkine (CX3CL1) is expressed as soluble or membrane-bound forms, with its actions being mediated through chemokine (C-X3-Cmotif) receptor 1 (fractalkine receptor) (CX3CR1). Fractalkine was upregulated on both CD4 and CD8 T lymphocytes in PAH [45] and it is likely that the increased expression of CX3CR1 on diseased PASMC contributes to the perivascular inflammatory cell influx and induce PASMC proliferation in MCT-induced PH [46].
Cytokines represent a large group of signaling proteins that are produced and secreted by cells of the immune system and regulate numerous biological processes including inflammation, immunity and hematopoiesis [47]. Inflammatory cytokines and chemokines seem to play a crucial role in the development of pulmonary hypertension. Cytokines emerged as major contributing factors in the pathogenesis of pulmonary hypertension as they can be used as biomarkers both for diagnosis and clinical outcome of patients with PH [44, 45, 48]. Here, we will review a few important common cytokines that have been well defined in clinical settings and studied experimental and transgenic animal models of PH like monocrotaline (MCT), Sugen + hypoxia and schistosoma-induced PH models. While these models cannot recapitulate human PAH, they provide a mechanistic insight into the connection between the host immune response and the pulmonary vascular disease, thus underscoring their utility as promising physiological surrogate of the human disease [4, 47].
IL-6 is an important signaling molecule and is produced by inflammatory cells, i.e. monocytes and T-lymphocytes. Studies have shown an increase in IL-6 in patients with PAH, and in rodents over-expression of IL-6 is adequate to cause experimental PH. Pharmacological blockade of IL-6 suppresses hypoxia-induced PH [49–51]. The IL6 – STAT3 – miR-17/92 – BMPR2 pathway play plausible role and may contribute in the pathogenesis of the pulmonary arterial remodeling [47]. Mice exposed to S. mansoni, as well as in the lung tissue of patients who died of this condition, showed a significant increase in IL-6/STAT3 signaling [52]. However, lack of IL-6 signaling by IL-6 genetic deficiency or with a pharmacologic STAT3 inhibitor resulted into more severe PH phenotype, including worse remodeling and RV hypertrophy [52]. This finding suggests that in mice exposed to Schistosoma, IL-6/STAT3 signaling is upregulated but in a compensatory manner, and that blockade of this pathway is detrimental.
Patients with PAH in association with connective tissue diseases showed higher IL-8 serum levels than patients without PAH and thereby play an important role in the development of PAH [53]. IL-8 is known to have proangiogenic and anti-apoptotic activities and acts as a growth factor for endothelial cells [54]. IL-8 might be involved in the hypoxic pressure response of pulmonary vessels as it was found elevated in early stages of high altitude pulmonary edema [55]. Similarly, IL-10 is also implicated in PAH as elevated levels were found in patients to counterregulate against inflammatory response in lung [47]. Further, injections of IL-10 reduced the mean pulmonary arterial pressure in MCT rats and significantly improved survival [56]. Overall, experimental data, however, suggest a protective role of the anti-apoptotic and anti-inflammatory cytokine IL-10.
The prototypic TH-2 cytokines IL-4 and IL-13 are critical in Schistosoma infection in the mouse [57]. One of the potential mechanisms underlying the pathogenesis of schistosomiasis-associated PAH is inflammation. IL-13 is a key inducer of several Type-2 cytokine-dependent pathologies. IL-13 regulates inflammation, mucus production, tissue remodeling, and fibrosis. IL-13Rα1 is the canonical IL-13 signaling receptor, whereas IL-13Rα2 is thought to be a competitive non-signaling decoy receptor. Other studies have shown that dysregulation of IL-13 signaling is present in human PAH; whether IL-13 is pro-proliferative or anti-proliferative remains unclear [30, 58, 59]. Favoring the pro-proliferative role(s) of IL-13, we found evidence of enhanced PH in S. mansoni-infected mice lacking IL-13Rα2, suggesting that IL-13 signaling is an important mediator of the granulomatous and vascular response to schistosomiasis infection [60]. Thus, IL-13 ligand and receptors may serve as novel biomarkers in PH and a potential target for receptor-directed biologic therapies.
Th1 and Th2 Immune Response Mediated Inflammatory Response in PAH
The effector responses of CD4+ T cells are generally divided into Th1, Th2, and Th17 responses; all there subsets appear to be involved in the pathogenesis of PH. In the Th1/Th17-skewed response, the arteriole may be invaded by mononuclear cells, including cytotoxic T cells, autoreactive B cells, autoantibodies, mast cells, and activated macrophages expressing granulocyte-macrophage colony-stimulating factor (GM-CSFR), inducible nitric oxide synthase (iNOS), and leukotriene B4 (LTB4). Th17 effector cells are induced in parallel to Th1 (producing interferon-γ, TNFs and IL-2), and, such as Th1, polarized Th17 cells have the capacity to cause inflammation and autoimmune disease. Both Th1 and Th17 colocalize regionally and may require each other for recruitment into the region [61]. Th17 cells not only produces IL-17 but it also produce cytokines like, IL-6, TNF-α, GM-CSF, IL-21, and IL-22. Th2 cells produce the cytokines IL-4, IL-5, and IL-13, which are involved in allergic responses and the clearance of extracellular worm driven antigens. When immune dysregulation favors TH1/TH17 immunity reactions, TNF-α and IL-6 seem harmful mediators promoting vascular remodeling [50, 62], whereas IL-6 may exert a protective effect in pulmonary vascular injury induced by Schistosomiasis [52]. In Th2-driven responses, there are unique inflammatory patterns. The inflammatory response is characterized by the recruitment of Th2 lymphocytes, mast cells, eosinophils and macrophages to the lung, and by elevated expression of allergen-specific immunoglobulin-E (IgE) in the serum [63]. It has been suggested that the chronicity of Th2 cytokine-mediated airway inflammation that is characteristic of allergic asthma is explained by the presence of a macrophage-like antigen-presenting cell population that persists in the airway lumen [64]. Further, transforming growth factor-β-mediated immunity response is closely linked with enhanced Th2 cytokines, e.g. IL-4 and IL-13 activity (Fig. 2.1), which then drives a destructive PH phenotype through pSTAT-6 signaling [65].
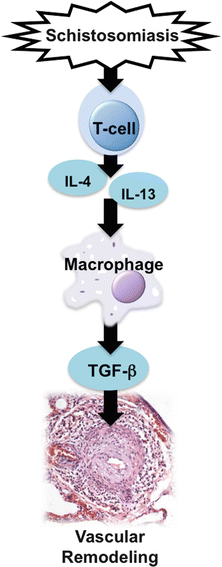
Fig. 2.1
Inflammatory pathways involved in the development of pulmonary arterial hypertension, highlighting the elucidated pathways triggered by Schistosoma infection
Macrophages and TGF-β Signaling in Pulmonary Vascular Remodeling
Among the inflammatory cells implicated in PAH, the monocyte/macrophage lineage has been consistently correlated with PH [31, 66–69]. However, macrophages efficiently respond to environmental signals with remarkable plasticity and undergo different forms of polarized activation that can be (simplistically) divided as classically activated (M1), alternatively activated (M2), and of an anti-inflammatory (regulatory) phenotype [70, 71]. Classically activated macrophages are effector phagocytes activated by interferon-γ and tumor necrosis factor (TNF-α). They produce inducible nitric oxide synthase and interleukin (IL)-12 and exhibit enhanced microbicidal or tumoricidal capacity [70]. On the other hand, M2-polarized macrophages are activated mostly by TH2 immune response mediated cytokines, IL-4 or IL-13 and express arginase-1 (Arg-1), found in inflammatory zone-1 (Fizz1), chitinase-3-like-3 (Ym1), and mannose receptor, C type lectin-1 [70–72]. M2 macrophages have been implicated in the pathogenesis of lung and other disorders via their ability to promote trophic, profibrotic, and angiogenic functions [73, 74]. The major characteristic of the third population, regulatory or anti-inflammatory macrophages, is the production of high IL-10 and low IL-12 levels and the promotion of immunosuppression [70, 72]. In the case of Schistosoma associated-PAH, once induced by Th2-CD4+ T-cells, macrophages stimulated by infection become alternatively activated (M2 phenotype) and, in the vascular adventitia, they can potentially represent the main cellular source of TGF-β (Fig. 2.1).
TGF-β signaling controls a plethora of cellular responses and is a member of a large family of multifunctional cytokines playing critical roles in embryogenesis, growth, wound repair, inflammation; TGF-β family has an important role in vascular homeostasis [75]. Abnormal TGF-β family signaling has been extensively linked to human PAH [76–78], and we and others have observed that blockade of TGF-β signaling suppresses PH due to monocrotaline or hypoxia in rodent models [65, 79, 80]. Recent studies have revealed significant insight into the mechanisms of the activation of TGF-β receptors through ligand binding, the activation of Smad proteins through phosphorylation, the transcriptional regulation of target gene expression, and the control of Smad protein activity and degradation [81, 82]. Recently, we found the Th2 cytokines IL-4 and IL-13 to be necessary for TGF-β activation in mice exposed to Schistosoma; previously, we observed IL-13 gain-of-function to be sufficient for TGF-β activation [60, 65]. We also found up-regulation of the TGF-β signaling pathway manifest by increased Smad2/3 phosphorylation in areas of vascular remodeling in both the mouse model and human tissue from subjects who died from schistosomiasis-associated PH [60, 65]. Schistosoma-induced PH phenotype in mice was partially suppressed by blockade at the level of the TGF-β ligand, type 1 receptor function, or the intracellular signaling molecule Smad3. Coupled with the finding of IL-4 and IL-13 suppression by TGF-β signaling blockade, there may be a positive feedback loop of IL-4/IL-13 and TGF-β propagating the disease [65]. Thus, another potential target for patients with PAH is blockade of TGF-β signaling.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


