Fig. 5.1
Excitation–contraction (EC) coupling in nonfailing a and failing b hearts. Adapted from Brum, PC et al. 2006, “Neurohumoral activation in heart failure: the role of adrenergic receptors”, Annals of the Brazilian Academy of Sciences 78(3): 485–503. a In nonfailing hearts during systole, EC coupling involves depolarization of the transverse tubule (T-tubule), which activates voltage-gated L-type Ca2 + channels (ICa) in the plasma membrane. Ca2 + influx via ICa triggers Ca2 + release from the sarcoplasmic reticulum (SR) via ryanodine receptors (RYR). During diastole, intracellular Ca2 + is pumped out of the cytoplasm by the SR Ca2 + ATPase (SERCA), which is regulated by phospholamban (PLB). In addition, Ca2 + is extruded from the cell by the sarcolemmal Na+/Ca2 + exchanger (NCX). The β-adrenergic receptor (β-AR) activation increases EC-coupling gain during systole and diastole through protein kinase A (PKA) phosphorylation of ICa, RYR, and PLB. The filled black arrows indicate the effects of aerobic exercise training. b In failing hearts, EC coupling is altered. RYR are hyperphosphorylated by PKA, which leads to greater sensitivity to Ca2 +-induced Ca2 + release at low and moderate cytoplasmic Ca2 + concentrations. The long-term effect of PKA hyperphosphorylation of RYR is an increased open probability at low intracellular Ca2 + concentrations, consistent with Ca2 + leakage during diastole. In addition, SERCA is downregulated, while NCX is upregulated in failing hearts, which contributes to depletion of SR Ca2 + stores. Aerobic exercise training attenuates abnormal Ca2 + handling, rescuing cardiac β1-AR to normal control levels and reducing β2-AR uncoupling. It also improves Ca2 + reuptake by SERCA, reduces sarcolemmal Ca2 + extrusion by NCX, and decreases Ca2 + leakage in diastole
Besides the autonomic balance of the cardiovascular system during a single bout of exercise, the aerobic exercise training is also able to modulate the density and responsiveness of β-AR in the heart. In general, cardiac β-AR density has been shown to decline with aerobic exercise training . However, data on such effects of exercise training are controversial in the literature [52, 53]. Taking into consideration the β-AR subtypes, studies have proposed a downregulation in either β1-AR or β2-AR subtypes after training [54–57]. This was suggested to be a compensatory adaptation in a tissue exposed to high concentrations of catecholamines during exercise training sessions [58]. Despite this response, compelling evidences from the literature suggest that aerobic exercise training increases cardiac contractility in animal models [59, 60] and humans [61]. In fact, increased adenylyl cyclase activity and myocardium responsiveness to β-AR agonists have been observed in trained rats regardless of training-induced alterations in cardiac structure [59]. Indeed, aerobic exercise training increases cardiac inotropy by phosphorylating key Ca2 + handling proteins, such as the RyR and phospholamban [62, 63] (Fig. 5.1a). In addition, an increased myofilament Ca2+ sensitivity and enhanced pH regulation are observed in isolated cardiac myocytes from exercise-trained rats [59, 60].
Cardiac Responses to Aerobic Exercise in HF
HF is a common endpoint of cardiovascular diseases, and the development of end-stage HF involves a continuous interaction between myocardial dysfunction and hyperactivation of neurohumoral systems, including adrenergic system. At first, this response is compensatory and may cause myocardium hypertrophy in response to increased cardiac work. However, sustained neurohumoral hyperactivity is toxic and deleterious as cardiac dysfunction persists [64]. Indeed, adrenergic system hyperactivation leads to worsening of HF, which is associated with altered cardiac adrenergic system components [65, 66], impaired Ca2 + handling [46, 48, 67, 68], and pathological cardiac remodeling [69, 70].
In failing hearts, there are abnormalities at multiple levels in the cardiac adrenergic signaling (Fig. 5.1b). β1-AR, the most expressed AR in the heart, is downregulated in HF with reduced responsiveness regardless of cardiomyopathy etiology [65, 71, 72]. In contrast, β2-AR levels remain unchanged in most studies [71, 72]. Additionally, the remaining cardiac β1– and β2-AR are desensitized mostly due to G-protein-coupled kinase-2 (GRK2) [72, 73], a kinase that phosphorylates and uncouples β-ARs. Increased GRK2-induced β-AR desensitization is supported by findings demonstrating that its inhibition reverses the pathological cardiac remodeling and improves cardiac function [72, 73]. The role of β3-AR in HF has not been elucidated yet, but it has been demonstrated that β3-AR signaling is increased in failing hearts [71].
Considering that adrenergic signaling in cardiac myocytes is tightly coupled to regulation of Ca2 + transients [74], desensitization of cardiac β-AR signaling pathway will lead to an abnormal Ca2 + homeostasis [46, 48]. In fact, altered expression and function of major Ca2+-regulating proteins have been described in severe HF models [75]. Reduced expression of sarcoplasmic reticulum Ca2+-ATPase and L-type Ca2 + channel paralleled by increased Ca2 + leaking by RyR and sarcolemmal Ca2 + extrusion by Na+/Ca2 + exchanger (NCX) is accounted for the reduced Ca2 + transient and depletion of sarcoplasmic reticulum Ca2 + content in HF [67] (Fig. 5.1b).
Cardiac remodeling associated with fibrosis and cardiac myocyte death is also a landmark of adrenergic system hyperactivity in HF, and the use of drugs that block neurohumoral hyperactivation, such as β-blockers and losartan (angiotensin II receptor antagonist), leads to an anticardiac remodeling associated with reduced mortality and improved cardiac function [68, 69, 76, 77].
Accumulated evidences have shown that aerobic exercise training is an efficient nonpharmacological strategy for HF therapy. Aerobic exercise training in HF patients improves quality of life, which is paralleled by an improved clinical symptom and a reduced hospitalization rate [78–80]. Additionally, beneficial effects of aerobic exercise training in HF are associated with a better autonomic control of the cardiovascular system [81, 82].
One of the first studies showing an improved autonomic balance after aerobic exercise training in HF individuals was published in 1992 by Coats and coworkers [83]. In fact, a landmark of aerobic exercise training in HF is a reduction in sympathetic hyperactivity [72, 84], which is paralleled by an improved cardiac Ca2 + handling [46, 48, 82, 85] and anticardiac remodeling [69, 82].
One of the mechanisms underlying the improved cardiac function by aerobic exercise training is related to attenuation of adrenergic signaling dysfunction in failing hearts associated with a rescue of cardiac β1-AR to a normal control level accompanied by increased cAMP and reduced GRK2 levels [86, 87]. The improved cardiac adrenergic signaling by aerobic exercise training in HF improves, at least in part, Ca2 + homeostasis. In fact, in a model of sympathetic hyperactivity-induced HF model in mice, Rolim and coworkers [46] have demonstrated that aerobic exercise training improved the net balance of cardiac Ca2 +-handling protein expression represented by improved Ca2 + reuptake by sarcoplasmic reticulum Ca2+-ATPase and reduced sarcolemmal Ca2 + extrusion by NCX. These results have been corroborated by other studies in different models of HF [59, 88]. The main effects of aerobic exercise training on intracellular Ca2 + handling proteins in HF are depicted in Fig. 5.1b.
Besides being an efficient therapy for HF, aerobic exercise training is considered an important strategy for the prevention of cardiovascular disease. Aerobic exercise training prior to HF development confers an important cardioprotector effect, attenuating cardiac dysfunction and abnormal Ca2 + handling [48, 89].
Regarding the impact of aerobic exercise training on cardiac remodeling induced by HF, reverse cardiac remodeling with a shift from pathological to physiological cardiac remodeling has been observed in exercise-trained animal models of HF [69, 90].
Despite structural similarities between physiological and pathological cardiac remodeling [90], studies have demonstrated that distinct molecular pathways are involved in each form of cardiac hypertrophy [60, 91–93]. While calcineurin/nuclear factor of activated T cell (NFAT) signaling pathway is one of the major players in pathological cardiac remodeling [60, 93], physiological cardiac remodeling is mainly associated with phosphatidylinositide-3-kinase/protein kinase B (PI3K/Akt) signaling cascade [91, 92]. In this sense, aerobic exercise training in animal models of HF has been implicated in both deactivation of pathological and activation of physiological pathways involved in cardiac remodeling. Thus, deactivaction of cardiac calcineurin/NFAT signaling pathway by aerobic exercise training has been observed in both sympathetic hyperactivity- and hypertrophic cardiomyopathy-induced HF in mice [69, 94]. Even though disruption of the AKT1 gene abrogates exercise training-induced physiological cardiac hypertrophy, PI3K/Akt pathways seem to play a subtle role in the cardiac antiremodeling effect induced by aerobic exercise training in HF [69, 95].
In summary, aerobic exercise training effectively attenuates the impaired cardiac adrenergic signaling and Ca2 + handling, and has a cardiac antiremodeling effect in failing hearts, which ultimately leads to an improved cardiac function in HF.
Circulatory Adjustments to Aerobic Exercise in Health and Disease: Role of Autonomic Nervous System
During exercise, circulatory adjustments are necessary to meet the metabolic demand of active skeletal muscle, and neural control of sympathetic nerve activity plays a major role in ensuring the efficacy of these adjustments [96], since parasympathetic vascular innervation is scarce [97] . In this sense, several neural mechanisms, working in concert, regulate sympathetic nerve activity to active (exercising muscle) and inactive (nonexercising muscle) vascular beds through complex interactions [98, 99]. Local mediators are also involved in this regulation [100].
It is worth to highlight that proper sympathetic nerve regulation to active and inactive vascular beds is of critical importance, since impaired vascular response to aerobic exercise in cardiovascular diseases (e.g., HF) is considered a main mechanism involved in exercise intolerance [101]. Moreover, reduced skeletal muscle blood flow is considered an independent predictor of mortality in patients with HF [102] (Fig. 5.2). In the following section, we will discuss the mechanisms (central and reflexes) that control the sympathetic activity and vascular conductance during exercise in normal subjects and in patients with HF.
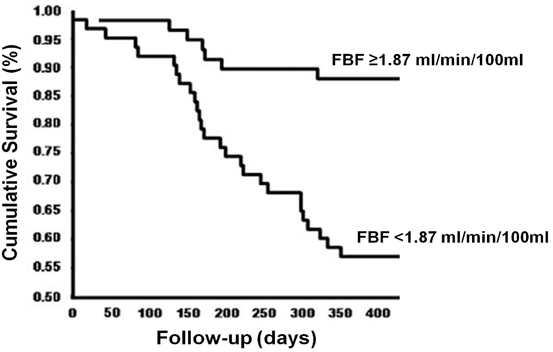
Fig. 5.2
Kaplan–Meier analysis of the cumulative rates of survival in patients with heart failure stratified in two groups on the basis of forearm blood flow (FBF, ml/min/100 ml). Survival rate was significantly lower in patients with forearm blood flow < 1.87 ml/min/100 ml (p = 0.002). Adapted from Barreto, ACP et al., 2009, “Increased muscle sympathetic nerve activity predicts mortality in heart failure patients,” International Journal of Cardiology 135(3): 302–307
Mechanisms Involved in Circulatory Responses to Aerobic Exercise
During exercise, a redistribution of cardiac output occurs to match the high metabolic need imposed by contracting muscle. In this sense, the regulation of sympathetic nervous system to different vascular beds during exercise is crucial to maintain and reset vascular conductance according to blood flow necessity, as well as, to preserve blood pressure and the perfusion of vital organs (e.g., brain and heart). The net result of this response is the increased vascular conductance and blood flow to active vascular beds while it decreases to inactive ones (e.g., liver, stomach, and skin) [103].
Activation of sympathetic nerve system during exercise is regulated by central command and reflex arising from skeletal muscle (exercise pressor reflex elicited by mechanoreceptors and metaboreceptors) or from aortic arch/carotid arteries (baroreceptors) [97, 103, 104].
At the onset of exercise, sympathoexcitation by central command increases its activity and induces the constriction of visceral and nonexercising muscle arterioles, metarterioles, and small and large veins. Besides its role in redirecting blood flow in favor of exercising muscles and central organs, sympathetic-mediated visceral vasoconstriction plays an important role in sustaining arterial pressure and maintaining an appropriate perfusion of vital organs [97, 103, 105]. Therefore, vasoconstriction of peripheral vessels is essential to the practice of exercise and it seems to be related to exercise intensity [106]. In exercising muscles, central command contributes to sympathoactivation only near maximal effort, which occurs mainly to counteract an exaggerated vasodilation, preventing blood pressure reduction and hypoperfusion, and consequently, a drop in muscle oxygen supply [22, 97, 107, 108].
Both central command and exercise pressor reflex exert an important role in resetting arterial baroreflex to higher blood pressure levels during exercise, which may further contribute to sympathoexcitation and simultaneous increase in heart rate and blood pressure during exercise [96, 104, 109, 110]. The resetting of arterial baroreflex operating point is paralleled by preserved baroreflex sensitivity during exercise [110–113], which seems to be very important to prevent excessive increase in blood pressure during exercise.
Concerning the cardiovascular response to exercise pressor reflex, metaboreflex elicited by afferent fiber type IV seems to have an important role in regulating sympathetic outflow to exercising and nonexercising vascular beds [97, 114–116]. Sympathoexcitation induced by metaboreflex was primarily thought to operate in moderate-to-high-intensity aerobic exercise when metabolites accumulate in a muscle undergoing contraction. However, metaboreflex is also elicited by a reduction in intracellular pH, which provides evidence for its activation even in mild exercise with minimal accumulation of muscle metabolites [24, 117–119]. Of interest, both sympathoexcitation [24, 117, 119] and increase in arterial blood pressure [118] display a significant inverse correlation with intracellular pH drop during exercise. Therefore, these findings give support for metaboreflex activation under inadequate blood flow to assure an appropriated blood supply to muscles’ and metabolites’ washout. Accordingly, metaboreflex seems to correct any mismatch between muscle blood flow and metabolism overriding central command .
As aforementioned, the main role of mechanoreflex elicited by afferent fibers type III is related to a cardiac vagal withdrawal contributing to exercise tachycardia at the onset of exercise [12]. Even though mechanoreflex contribution for sympathetic activation during muscle contraction has been demonstrated in animals [25, 120], it is difficult to isolate its contribution for sympathoexcitation in humans [12]. This is mainly due to the fact that subpopulations of afferent type III fibers are polymodal, being sensitized by either metabolites or mechanical stimulus [23, 121]. Indeed, it has been demonstrated that muscle mechanoreceptor is sensitized by metabolites [122].
Even though central and reflex activation of sympathetic nerves leads to vasoconstrictor stimulus in active and inactive vascular beds, an increased vascular conductance is observed in exercising muscles. This is due to local vasodilator mechanisms (metabolic vasodilation, shear-stress induced vasodilation, muscular pump, etc), which surpass the vasoconstrictor stimulus matching the high metabolic demand in exercising muscles. Indeed, a reduced responsiveness of active vascular beds to vasoconstrictor stimulus has long been reported [107, 123, 124]. In fact, Remensnyder and coworkers [123] coined the term “functional sympatholysis” to describe this phenomenon. The mechanisms proposed to explain functional sympatholysis include either prejunctional inhibition of noradrenaline release or postjuctional inhibition of noradrenaline binding to adrenergic receptors , which is mediated by metabolites produced in contracting muscles [97]. However, the precise mechanism underlying functional sympatholysis, or even its existence, is currently under debate [125, 126].
Circulatory Adjustments to Acute and Chronic Exercise in Heart Failure
Sympathoexcitation is a well-documented feature of chronic HF, leading to an increased vasoconstrictor tone and reduced blood flow to muscles and other organs. For instance, a consequence of reduced renal blood supply is an increased renin secretion and inappropriate salt and water retention. In fact, HF patients display lower renal cortical and skeletal muscle vascular conductance than healthy individuals [127]. These responses are of clinical importance since impaired exercise-mediated vasodilation by increased circulating noradrenaline is considered independent predictors of mortality in HF patients [102, 128]. Indeed, reduced skeletal muscle blood flow is a main mechanism involved in exercise intolerance in HF [101, 129].
As aforementioned, increased sympathetic activity to exercising vascular beds is offset by local mechanisms, such as increased vasodilation induced by metabolic byproducts. However, sympathetic hyperactivity observed in HF patients changes the balance between vasodilation and vasoconstriction in favor of the former. In fact, the reflex forearm vasodilatory response to handgrip contraction is restored under intra-arterial α-adrenergic receptor blockade by phentolamine in HF patients without affecting arterial pressure [130]. In contrast, reflex vasodilatory forearm blood flow in response to mental stress is not affected by either intra-arterial infusion of acetylcholine or l-arginine [131].
The beneficial effects of aerobic exercise training in HF include a significant reduction in sympathetic hyperactivity [48, 76, 84, 132], as illustrated in Fig. 5.3. In fact, Coats and coworkers [83] were the first to demonstrate that aerobic exercise training reduced by 16 % whole-body radiolabeled norepinephrine spillover in HF patients parallel by an improved cardiac autonomic balance. This is particularly interesting since one of the main pharmacological therapies for HF is the use of β-AR blockade. Therefore, HF therapies that decrease adrenergic hyperactivity should combine aerobic exercise training to optimize the benefits on the cardiovascular system . In this context, we have demonstrated an additional reduction in muscular sympathetic nerve activity by aerobic exercise training in HF patients optimized with carvedilol, a third-generation β-blocker (β1– and β2-blockade) with an α-blockade and vasodilatory effect [133]. Additionally, while β-blocker therapy has no impact on exercise capacity in humans or animals with HF [77, 134], a β-blocker combined with exercise training improves exercise capacity [48, 68, 133]. Indeed, it is important to highlight that reduction in sympathetic hyperactivity by exercise training in HF patients is associated with a better clinical outcome [84, 133], which occurs independently of HF patient age [135] or gender [136].
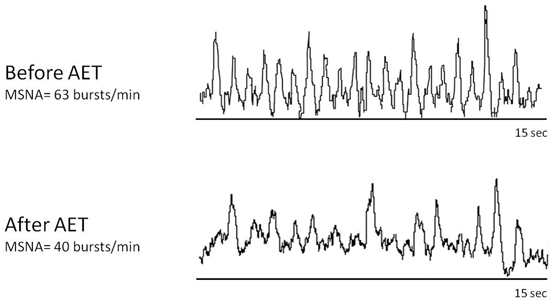
Fig. 5.3
Muscle sympathetic nerve activity (MSNA) of a 58-year-old female heart failure patient (hypertensive cardiomyopathy) with a 36 % ejection fraction before and after 4 months of aerobic exercise training (AET). MSNA was obtained by direct recording of muscle sympathetic discharge by microneurography. Note that AET dramatically reduced sympathetic discharge. Data are from Cardiovascular Rehabilitation and Exercise Physiology Laboratory, Heart Institute, Medical School, Universidad de São Paulo
Potential mechanisms underlying reduced sympathetic nerve activity by aerobic exercise training involve the afferent autonomic control of sympathetic nerve activity coordinated by arterial baroreceptors, mechanoreceptors, and metaboreceptors [112, 113, 132, 137, 138].
Exaggerated sympathetic activation by central command is observed in HF. In fact, Koba and coworkers [129] demonstrated that renal and lumbar sympathetic nerve responses to mesencephalic locomotor region stimulation were exaggerated in myocardial infarcted animals. Likewise, increased exercise pressor reflex has been observed in HF [20, 138]. Mechanoreflex overactivity is involved in exaggerated exercise pressor reflex in HF, while the role played by metaboreceptor is still under debate, since some studies suggest a blunted metaboreflex function in HF [139–143] and others demonstrate a metaboreflex overactivation in HF [138, 144, 145]. Of interest, aerobic exercise training attenuates the overactive exercise pressor reflex in HF related mainly through reduced mechanoreflex evidenced by studies conducted in either animals or humans [138, 139].
Increased sympathetic nerve activity in HF is also associated with reduced baroreflex sensitivity in animals [146, 147] and humans [148, 149]. This is of particular interest since impaired baroreflex sensitivity is associated with a poor prognosis in HF patients [150–152]. Regarding the effect of aerobic exercise training on baroreflex function, our group demonstrated that aerobic exercise training reduced renal sympathetic nerve activity associated with an increased arterial baroreceptor afferent sensitivity in control rats [113, 137, 153]. Interestingly, this knowledge was extended to HF by Liu and coworkers [132] who observed reestablished arterial baroreflex control to renal sympathetic nerve activity by aerobic exercise training in a rabbit model of pacing-induced HF. In addition, Rondon and coworkers [154] also demonstrated that improved baroreflex control of renal sympathetic nerve activity in myocardial infarcted rats was associated with increased aortic depressor nerve sensitivity. Importantly, aerobic exercise training improved arterial baroreflex sensitivity in HF patients [155, 156]. The clinical relevance of these findings was firstly demonstrated by La Rovere and coworkers [152], who observed that exercise training-induced increase in baroreflex sensitivity was able to predict improved prognosis in myocardial infarction patients.
In summary, aerobic exercise training reduces sympathetic hyperactivity in HF, which is associated with an improved outcome. The mechanisms underlying this response involve central and reflex adjustments of adrenergic system that regulate cardiovascular function.
References
1.
Kokkinos P, Myers J. Exercise and physical activity: clinical outcomes and applications. Circulation. 2010;122(16):1637–48.PubMed
2.
WHO. Global health risks: mortality and burden of disease attributable to selected major risks. Geneva: World Health Organization; 2009.
3.
WHO. Global recommendations on physical activity for health. Geneva: World Health Organization; 2010.
4.
Hallal PC, Andersen LB, Bull FC, Guthold R, Haskell W, Ekelund U, et al. Global physical activity levels: surveillance progress, pitfalls, and prospects. Lancet. 2012;380(9838):247–57.PubMed
5.
Booth FW, Lees SJ. Fundamental questions about genes, inactivity, and chronic diseases. Physiol Genomics. 2007;28(2):146–57.PubMed
6.
Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346(11):793–801.PubMed
7.
Garber CE, Blissmer B, Deschenes MR, Franklin BA, Lamonte MJ, Lee IM, et al. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334–59.PubMed
8.
Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17(2):162–84.PubMed
9.
Tipton CM. History of exercise physiology. Champaigh: Human Kinetics; 2014.
10.
Brum PC, Bacurau AV, Cunha TF, Bechara LR, Moreira JB. Skeletal myopathy in heart failure: effects of aerobic exercise training. Exp Physiol. 2014;99(4):616–20.PubMed
11.
von Haehling S Steinbeck L Doehner W Springer J Anker SD. Muscle wasting in heart failure: An overview. Int J Biochem Cell Biol. 2013;45(10):2257–65.
12.
Nobrega AC, O’Leary D, Silva BM, Marongiu E, Piepoli MF, Crisafulli A. Neural regulation of cardiovascular response to exercise: role of central command and peripheral afferents. Biomed Res Int. 2014;2014:478965.PubMedCentralPubMed
13.
Lymperopoulos A. Physiology and pharmacology of the cardiovascular adrenergic system. Front Physiol. 2013;4:240.PubMedCentralPubMed
14.
Goodwin GM, McCloskey DI, Mitchell JH. Cardiovascular and respiratory responses to changes in central command during isometric exercise at constant muscle tension. J Physiol. 1972;226(1):173–90.PubMedCentralPubMed
15.
Mitchell JH. Neural control of the circulation during exercise: insights from the 1970–1971 Oxford studies. Exp Physiol. 2012;97(1):14–9.PubMed
16.
Strange S, Secher NH, Pawelczyk JA, Karpakka J, Christensen NJ, Mitchell JH, et al. Neural control of cardiovascular responses and of ventilation during dynamic exercise in man. J Physiol. 1993;470:693–704.PubMedCentralPubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


