The Cardiomyopathic and Inflammatory Diseases
Jeffrey David Hosenpud
Bart L. Cox
The concept of primary myocardial disease was first suggested in the late nineteenth century by Krehl, who described several cases of “chronic myocarditis” in which autopsied hearts demonstrated myocardial degeneration, hypertrophy, and inflammation (1). The term primary myocardial disease was coined by Josserand and Galla-vardin at the beginning of this century (2). However, it was not until 1957 that the term cardiomyopathy was introduced by Brigden to describe myocardial disease of unknown etiology occurring in the absence of coronary disease (3). Subsequently, in 1968, the terminology was specifically defined by the World Health Organization as myocardial disease excluding vascular, hypertensive, and valvular disease. Cardiomyopathy was further subclassified as primary or secondary, depending on whether its causes were unknown or known (4). The definition was further refined in 1980 by a combined task force of the World Health Organization and the International Society and Federation of Cardiology, which exchanged the term secondary cardiomyopathy for specific heart-muscle disease (5). Despite these very specific definitions, the term cardiomyopathy is used by most to denote primary myocardial disease, either of specific or unknown etiology. Unfortunately, it is used by many as a synonym for ventricular dysfunaion of any cause (e.g., ischemic cardiomyopathy. valvular cardiomyopathy). For the purposes of this chapter, the term cardiomyopathy will be used to denote diseases of the myocardium of either known or unknown etiology.
Traditionally, the cardiomyopathies have been grouped according to anatomical-physiological rather than etiological features These categories include dilated, hypertrophic, and restrictive cardiomyopathy. The dilated form is most common, and the restrictive form least common. Although this classification does not take into account underlying etiologies, more often than not specific etiologies tend to fall into specific anatomical groupings. Furthermore, this anatomical classification is quite helpful in the medical management of patients with cardiomyopathy, as is described later. Finally, prognosis is more closely linked to anatomical-physiological class rather than specific etiology in most cases.
Dilated Cardiomyopathy
Pathophysiology and Mechanisms of Disease
The primary myocardial defect in patients with dilated cardiomyopathy is an abnormality in contraction. As a result of reduction in myocardial contractility, the heart dilates out of proportion to the degree of hypertrophy, resulting in four-chamber enlargement with relatively thin cardiac walls (6). This increase in radius relative to wall thickness increases myocardial wall stress (7) and decreases subendocardial coronary flow (8), leading to further cardiac dysfunction. Figure 18-1 demonstrates a typical echocar-diogram from a patient with dilated cardiomyopathy. The M-mode echocardiogram shows the left ventricle (LV) to be markedly enlarged and systolic function reduced. The two-dimensional echocardiogram from the same patient demonstrates that all four chambers are enlarged.
In most cases of dilated cardiomyopathy, the mech-anism(s) for the initial myocardial insult are unknown. Furthermore, it is unclear whether the defect is one of primary individual myocardial cell loss or cell dysfunction followed ultimately by myocardial cell loss and
replacement with fibrous connective tissue. A host of molecular, biochemical, and cellular abnormalities have been described in patients with dilated cardiomyopathy. These include alterations in calcium handling by the sarcoplasmic reticulum (9), a reduction β1-receptor density (10,11), alterations in Gs and Gi proteins (12,13), alterations in cholinergic receptor density and function (14), and expression of altered or fetal myosin subtypes (15). It is not clear, however, whether any of these noted abnormalities is specific to dilated cardiomyopathy or is a more general manifestation of cardiac dilatation and failure.
replacement with fibrous connective tissue. A host of molecular, biochemical, and cellular abnormalities have been described in patients with dilated cardiomyopathy. These include alterations in calcium handling by the sarcoplasmic reticulum (9), a reduction β1-receptor density (10,11), alterations in Gs and Gi proteins (12,13), alterations in cholinergic receptor density and function (14), and expression of altered or fetal myosin subtypes (15). It is not clear, however, whether any of these noted abnormalities is specific to dilated cardiomyopathy or is a more general manifestation of cardiac dilatation and failure.
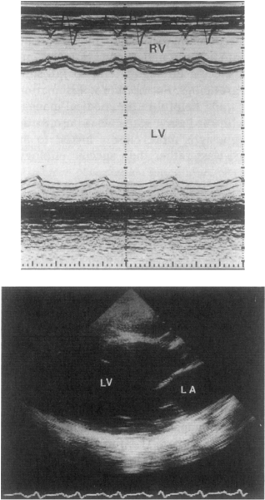 Figure 18-1 An M-mode (top) and two-dimensional (bottom) echocardiogram from a patient with dilated cardiomyopathy. Note the large chamber sizes and poor systolic contraction. LV, left ventricle; RV, right ventricle; LA, left atrium. |
As previously noted, as early as the late nineteenth century it was hypothesized that cardiomyopathy is the result of chronic inflammatory disease (1). About 80 years later, the concept that dilated cardiomyopathy might be the result of an acute, subacute, or chronic myocarditis again received attention (16). The hypothesis put forth was that a myocardial insult, be it an acute viral infection or toxin, somehow altered the antigenicity of the myocardium so that it was mistaken for foreign tissue and an autoimmune attack was elicited (17). The problem with this hypothesis is that the pieces of the puzzle have been difficult to prove in a convincing way despite major research efforts to support them.
In terms of the initial insult, it has long been appreciated that the coxsackie group B virus is an important pathogen in acute viral myocarditis, and this has provided an excellent model for the study of acute myocarditis in animals. Furthermore, in some murine strains, acute cox-sackievirus myocarditis leads to a more chronic myocarditis and cardiac dilation suggestive of dilated cardiomyopathy (18). The difficulty in linking coxsackievirus to human dilated cardiomyopathy has been the inability to identify the virus in cardiac tissue. Serology is nonspecific (19), viral cultures are rarely positive (20), and electron microscopy for viral particles is difficult to interpret. With the recent growth of molecular biological techniques, new methods for viral diagnosis have become available. Bowles et al. (21) utilized in vitro nucleic acid hybridization to study whether enterovirus RNA is present in the myocardium of patients with dilated cardiomyopathy (21). Of the 40 patients studied, 21 had dilated cardiomyopathy and 19 were controls who had heart failure secondary to either coronary disease or congenital heart disease. Enterovirus RNA was detected in the myocardium of 6 of the 21 patients with dilated cardiomyopathy and only one of the controls.
A number of studies have investigated the immunological milieu in dilated cardiomyopathy. Limas and Limas (22) found that HLA-DR4 was represented in 40% of patients with dilated cardiomyopathy, but in only 24% of controls. Koike (23) investigated cell-mediated immune function in 18 patients with dilated cardiomyopathy. In these patients, phytohemagglutinin (PHA)-mediated lymphocyte blastogenesis was reduced; the ratio of CD4+ (helper) to CD8+ (suppressor) lymphocytes was higher; the PHA-mediated release of interleukin-2 (IL-2), a T-cell growth factor, was enhanced; and the number of activated lymphocytes (positive for IL-2 receptor) was reduced in comparison with controls. These data would suggest an inherited predisposition to immunological disease.
Antimyocardial antibodies have been extensively studied during the past several years. However, a link between the presence of antibody and an alteration in cardiac function or metabolism has not been established. In previous studies by Schultheiss et al. (24), an antibody directed against the adenosine diphosphate-adenosine triphos-phate (ADP-ATP) carrier protein of the mitochondrial membrane was described in patients with dilated cardiomyopathy. The regulation of β1-receptors on myocardium in patients with heart failure has been well-described (10). The mechanism for this downregulation is felt to be a negative feedback related to chronically elevated levels of circulating catecholamines, presumably a nonspecific mechanism related to the degree of heart failure. Limas et al. (25) demonstrated that a high percentage of patients with dilated cardiomyopathy, especially those in whom HLA-DR4 was represented, had circulating anti-β1-receptor autoantibodies. An additional mechanism for modulating cell surface β-receptors specific to dilated cardiomyopathy could therefore be hypothesized.
Mechanisms for the development of dilated cardiomy-opathy other than those involving immunological abnormalities have been investigated. Based on their experimental data derived from the Syrian hamster, Factor et al. (27) postulated that some forms of dilated cardiomyopathy are secondary
to small-vessel disease and specifically to microvascular spasm. This mechanism is presumed to play an important role in the dilated cardiomyopathy associated with pheochromocytoma (28). A study by Treasure et al. (29) demonstrated that the endothelium-dependent dilation of the coronary microvasculature is impaired in patients who have dilated cardiomyopathy in comparison with controls. Van Hoeven and Factor (30) reviewed endomyocardial biopsy specimens from 145 patients with dilated cardiomyopathy and demonstrated small-vessel thickening in 56% of these patients. This was not a specific finding, however, as a significant proportion of patients with hypertension, diabetes, unexplained arrhythmias, and chest pain syndromes had similar changes.
to small-vessel disease and specifically to microvascular spasm. This mechanism is presumed to play an important role in the dilated cardiomyopathy associated with pheochromocytoma (28). A study by Treasure et al. (29) demonstrated that the endothelium-dependent dilation of the coronary microvasculature is impaired in patients who have dilated cardiomyopathy in comparison with controls. Van Hoeven and Factor (30) reviewed endomyocardial biopsy specimens from 145 patients with dilated cardiomyopathy and demonstrated small-vessel thickening in 56% of these patients. This was not a specific finding, however, as a significant proportion of patients with hypertension, diabetes, unexplained arrhythmias, and chest pain syndromes had similar changes.
Direct myocardial toxicity is suggested for alcohol-induced cardiomyopathy (31) and is clearly present in anthracycline-induced dilated cardiomyopathy (31,32,33). Although alcohol is a potent depressant of myocardial function in the muscle bath, the variable effects of alcohol on the myocardium in vivo are poorly understood. Although several mechanisms (to be discussed later) have been suggested for anthracydine cardiotoxicity, there is at least a dose-related effect of anthracyclines on cardiac function.
What is becoming increasingly clear is that many of the idiopathic dilated cardiomyopathies have a genetic basis and, with more extensive screening, appear to be familial. This is a fairly new concept as, historically, most considered dilated cardiomyopathy to be sporadic in nature, unlike well-defined genetic diseases such as hypertrophic cardiomyopathy, which will be discussed later in this chapter. In fact, familial disease was found in as high as 48% of patients with apparent idiopathic cardiomyopathy when cardiac enlargement by echocardiography was used as a marker for early cardiomyopathy (34). Other studies using echo and other screening have suggested prevalences in the 25% to 35% range depending on the criteria used (35,36).
A recent review of the subject of familial dilated cardiomyopathy discusses these prior investigations as well as the specific genetic abnormalities identified to date. Ninety percent of familial dilated cardiomyopathy appear to be inherited with autosomal dominance, with 16 gene mutations identified. Defects in the lamin A/C or β-myosin heavy chain account for between 5% to 10%, with abnormalities in actin and desmin being less common (37). X-linked familial cardiomyopathy usually secondary to defects in the dystrophin gene may account for between 5% and 10% of familial dilated cardiomyopathy and is usually also associated with skeletal muscle involvement (muscular dystrophies). Rarely, autosomal recessive inheritance patterns have been described, as in one family with a mutation in the cardiac troponin I gene (37).
In most cases, proof of a genetic cause of a cardiomyopathy has a limited impact on the treatment of the index patient. Gene therapy, referring to the introduction of altered or foreign genes into human tissues for the purpose of correcting a genetic defect, is not now feasible for most genetic cardiomyopathies, and there are serious logistic reasons to question whether such treatment will ever be possible or desirable. However, the demonstration that a cardiomyopathy is genetic in origin has important implications beyond those of clinical management, such as the need for family screening to identify other affected persons, or for genetic counseling of affected patients who wish to have children.
Specific Causes or Associations and Dilated Cardiomyopathy
Table 18-1 provides a partial listing of the causes of and conditions associated with dilated cardiomyopathy. Although the list is extensive, in the vast majority of patients there will be no obvious cause of disease
Duchenne and Becker Muscular Dystrophy, and X-Linked Cardiomyopathy
Duchenne muscular dystrophy, described in 1868 as a “hypertrophic paraplegia of infancy” (38), is an X-linked recessive neuromuscular disorder that affects primarily boys. Clinical manifestations are usually evident by age 5. Progressive muscle weakness is usually first evident in the lower extremities, with involvement of proximal musdes being more marked than that of distal musdes. Calf musdes, and sometimes other muscles as well, become enlarged, partly owing to true muscle hypertrophy and partly because of infiltration of the musde with fat. Joint contractures develop and walking becomes more and more difficult. Respiratory musde weakness leads to pulmonary infection and respiratory failure. Intellectual impairment may also occur. Laboratory findings include elevation of creatine kiriase and; electromyographic abnormalities (39).
Genetic mapping techniques led in 1986 to the doning and identification of the gene for Duchenne muscular dystrophy (40,41). As previously noted, the product of this gene, called dystrophin (42), is a rod-shaped cytoskeletal protein that is normally found on the cytoplasmic surface of the sarcolemma (43,44). It is known to bind on its amino-terminal end to actin (45) and on its carboxy-terminal end to a membrane-spanning group of proteins called the dystrophin-assodated glycoprotein complex (46,47). One
of the extracellular components of this complex named α-dystroglycan, binds to the extracellular matrix protein laminin (48). The result is that dystrophin acts as a link between the cytoskeleton and the extracellular matrix (49). It is therefore theorized that the function of dystrophin is to stabilize the sarcolemma and to protect it from the mechanical stresses of muscle contraction (50).
of the extracellular components of this complex named α-dystroglycan, binds to the extracellular matrix protein laminin (48). The result is that dystrophin acts as a link between the cytoskeleton and the extracellular matrix (49). It is therefore theorized that the function of dystrophin is to stabilize the sarcolemma and to protect it from the mechanical stresses of muscle contraction (50).
Table 18-1 Reported Causes of and Associations with Dilated Cardiomyopathy | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In patients with Duchenne muscular dystrophy, dystrophin is almost entirely absent from the muscle cell membrane (51,52). The absence of dystrophin from the sarcolemma is thought to expose the membrane to mechanical stress, which leads to abnormal ion fluxes, loss of force transduction across the membrane, and perhaps disruption of the membrane itself (53,54,55). Genetic studies have demonstrated that a variety of mutations, including deletions, duplications, and point mutations (56,57,58), can all lead to dystrophin deficiency.
Cardiac involvement has long been recognized to be a feature of Duchenne muscular dystrophy (59,60). Electrocardiograms are often abnormal in childhood (61) and echocardiography demonstrates that LV dysfunction is the rule by age 18 (62,63). Symptomatic congestive heart failure is less common, partly because the patient’s progressive motor debility places decreasing demands on cardiac function (64).
The extent of cardiac involvement does not correlate well with the severity of the skeletal muscle disorder (65). The myocardial dysfunction is most marked in the poster-obasal LV free wall (66,67); not only is this region of the heart hypokinetic but also its echodensity often appears to be increased, suggestive of fibrosis (68). These observations have led to the hypothesis that myofiber orientation and force distribution in the posterior LV differ from those of the anterior wall, leading to greater and earlier damage to the sarcolemma in the posterior wall in the absence of dystrophin (69). The focal nature of the ventricular dysfunction seen on noninvasive imaging corresponds to the histopaihological findings, which include myofibrillar loss and fibrosis most pronounced in the posterobasal LV (70,71). These observations may also explain the typical electrocardiographic features, which include tall R waves in leads V1 and V2 and deep, narrow Q waves in the lateral and sometimes inferior leads (72). Although dilated car-diomyopathy is the most common form of disease seen in Duchenne muscular dystrophy, hypertrophic cardiomy-opathy is seen in some patients (73). Arrhythmias and conduction disturbances have also been described (74). Because cardiac involvement may contribute to morbidity and may at times be the cause of death, regular screening for cardiac manifestations is recommended (75).
Becker muscular dystrophy is a less-severe form of X-linked muscular dystrophy. The clinical manifestations are similar to those of Duchenne muscular dystrophy but they occur later in life and progress more slowly (76,77). With the identification of the Duchenne muscular dystrophy gene and its protein product, it has been demonstrated that Becker muscular dystrophy is also a consequence of mutations in dystrophin (78,79). Typically, mutations causing Becker muscular dystrophy result in the production of a structurally abnormal dystrophin protein; in contrast, mutations causing Duchenne muscular dystrophy result in a severe quantitative deficiency of the protein (80,81). The clinical Collrse and degree of cardiac involve-ment in Becker muscular dystrophy are much more van-able than in Duchenne muscular dystrophy (82,83). Data suggest that even among sibling pairs with identical dystrophin mutations, the onset of cardiomyopathy is variable and unpredictable (84), and the cardiac dysfunction may precede the evidence of skeletal muscle weakness (85). Cardiac failure is actually a more common cause of death in Becker muscular dystrophy than in Duchenne muscular dystrophy. This may be because the skeletal muscle function of patients with Becker muscular dystrophy is relatively well-preserved, so that they remain more active and place a heavier work load on the myocardium, which leads to a more rapid progression of cardiomyopathy (86,87).
In 1993, Towbin et al. (88) reported that the family originally described by Berko and Swift, as well as another family with X-linked dilated cardiomyopathy, showed evidence of genetic linkage to the 5′ portion of the dystrophin gene locus. This report provided the first evidence that a dilated cardiomyopathy without any skeletal muscle manifestations Collld be caused by an abnormality of dystrophin. Since then, numerous other families with dystrophin mutations and isolated dilated cardiomyopathy have been described (89,90,91,92). Some of these reports have also investigated the abundance and structure of dystrophin in the heart and skeletal muscle of the subject families; both qualitative (93) and quantitative (94) dystrophin abnormalities have been detected. Absence of cardiac membrane-associated dystrophin can be seen with normal skeletal muscle findings (95,96).
A family history in which only the male relatives have dilated cardiomyopathy may be the first clue that a patient has a dystrophinopathy; however, some women may be affected as manifesting carriers, as previously noted (97). Furthermore, as many as a third of cases of Duchenne muscular dystrophy represent de novo mutations with no family history (98). Therefore, a diagnosis may be difficult to make based on kindred analysis alone. Creatine kinase levels are almost always elevated in patients with a dystrophin disorder; such elevations should prompt further investigation of skeletal muscle function, possibly a cardiac and skeletal muscle biopsy, and consideration of dystrophin gene or protein analysis (99).
Other Muscular Dystrophies
Several other muscular dystrophies are known to have a significantly frequent association with cardiac involvement. Emery-Dreifuss muscular dystrophy is an X-liriked disorder that, in contrast to Duchenne muscular dystrophy, is characterized by an absence of calf hypertrophy and a distinct pattern of muscle wasting referred to as humeroperoneal (proximal muscles of the upper extremities and distal muscles of the lower extremities are the most affected) (100,101). Joint contractures are a prominent feature of this condition, especially at the elbows, Achilles tendons, and posterior cervical muscles (102). The most common forms of cardiac involvement are rhythm disturbances, including atrial fibrillation or flutter and conduction block (103). However, dilated cardiomyopathy has also been described (104,105) and cardiac transplantation for cardiac failure has been reported (106). Genetic mapping of the
relevant region of the X chromosome has led to the identification of the responsible gene (107). The protein product of this gene has been named emerin. It has been demonstrated to localize to the inner membrane of the nucleus (108) but its function has not yet been determined. A disorder that is clinically indistinguishable from Emery-Dreifuss muscular dystrophy, but with an autosomal dominant mode of inheritance, has also been described (109).
relevant region of the X chromosome has led to the identification of the responsible gene (107). The protein product of this gene has been named emerin. It has been demonstrated to localize to the inner membrane of the nucleus (108) but its function has not yet been determined. A disorder that is clinically indistinguishable from Emery-Dreifuss muscular dystrophy, but with an autosomal dominant mode of inheritance, has also been described (109).
Limb-girdle muscular dystrophy refers to a group of disorders, some with autosomal dominant and some with autosomal recessive transmission, that typically are characterized by proximal muscle weakness and wasting that progresses to involve all extremiuies; the face is spared (110). Conduction system disease has been described in limbgirdle muscular dystrophy type I, and dilated cardiomy-opathy has been reported (111,112,113).
Mitochondrial Myopathies
The mitochondria are the site of oxidative phosphorylation, the metabolic process that generates most of the ATP produced by eukaryotic cells. Pyruvic acid, the product of glycolysis, is taken up into the mitochondrial matrix and reacts with coenzyme A (CoA) to form acetyl CoA. The enzymes of the citric acid cycle, mostly in solution in the mitochondrial matrix, oxidize the acetyl group of acetyl CoA to carbon dioxide and, in the process, reduce nicoti-namide adenine dinucleotide (NAD) to NADH and flavin adenine dinucleotide (FAD) to FADH2. Protein complexes on the inner mitochondrial membrane then transport electrons from NADH and FADH2 through the cytochrome system to oxygen. The energy accumulated during this process is used to create high-energy phosphate bonds, which can then be used for all the metabolic requirements of the cell (114). Muscle and nerve cells, which rely heavily on oxidative metabolism to supply their high energy demands, typically contain large numbers of mitochondria (115). Genetic abnormalities of mitochondrial function, especially of oxidative phosphorylation, therefore tend to affect the nervous system, skeletal muscle, and the heart, although they may affect a wide variety of other organ systems as well (116). Mitochondria have their own genome, a circular molecule of DNA that is distinct from the nuclear chromosome. Mitochondrial DNA encodes several of the component polypeptides of the enzymatic pathway for oxidative phosphorylation; in addition, it encodes a set of distinct mitochondrial ribosomal RNAs and transfer RNAs, which are necessary for translation of the mitochondrial genes (117). The replication, transcription, and translation of mitochondrial DNA occur independently of the analogous nuclear processes. Each mitochondrion typically contains several copies of the mitochondrial genome, and each cell usually contains numerous mitochondria. Thus, the stoichiometry of mitochondrial genetic information is quite different from the strict diploidy of nuclear genes. Mutations may occur in one or several copies of the mitochondrial DNA but not in others. When cells divide, the daughter cells may inherit unequal numbers of mitochondria, some bearing mutations and others identical to the germline (118).
Mitochondrial DNA is maternally inherited—children inherit virtually all their mitochondrial genes from their mother, not their father (119). This is a consequence of the fact that the great majority of the mitochondria in a developing embryo are acquired from the ovum; the sperm contains very few mitochondria. As a result, mutations of mitochondrial DNA are transmitted from mother to child, whereas paternal mitochondrial mutations are not transmitted (120). This maternal pattern of inheritance is distinct from the usual mendelian categories and may be the first evidence leading to clinical recognition of a mitochondrial disorder (121,122). However, because some of the enzymatic components involved in oxidative phosphorylation are encoded in the nucleus, genetic defects of mitochondrial function may also exhibit mendelian inheritance (123). Furthermore, abnormalities of mitochondrial DNA have been identified that exhibit autosomal dominant inheritance, which suggests that nuclear genes responsible for the regulation of mitochondrial DNA synthesis are affected (124,125). Finally, because mitochondrial DNA has an exceptionally high mutation rate, many cases of mitochondrial disease occur in the absence of any family history (126).
The diseases of oxidative phosphorylation are clinically diverse, with heterogenous manifestations. There is a wide variety of possible mutations, induding point mutations, deletions, insertions, and multiple sequence anomalies. These genetic alterations may be transmitted in varying copy numbers to different tissues and organs, and the requirements of the tissues and organs themselves for the energy produced by oxidative metabolism may vary. Over time, additional mutations may accumulate in the mitochondria of some tissues but not others (127,128). As a result, it is difficult to establish a useful dassification system for mitochondrial diseases (129,130). Nonetheless, several distinct phenotypes have been identified and are used in describing individual patients with disorders of oxidative phosphorylation (131,132,133).
An example of this is the Keams-Sayre syndrome, which was first described in 1958 (134). The diagnosis is defined by onset before the age of 20, external ophthalmoplegia, ptosis, and pigmentary retinopathy. In addition, a number of other clinical features may be present including heart block, skeletal musde weakness, cerebellar dysfunction, deafness, and elevated cerebrospinal fluid protein (135,136,137). Conduction disturbances in Kearns-Sayre syndrome include left anterior hemiblock, right bundle-branch block, Mobitz type II second-degree atrioventricular block, and complete heart block (138,139). Permanent pacemakers have prolonged survival significantly in Kearns-Sayre syndrome type II. Dilated cardiomyopathy has been reported in Kearns-Sayre syndrome (140,141) and patients have undergone successful cardiac transplantation (142,143).
On skeletal muscle biopsy in Kearns-Sayre syndrome, myocytes stained with the Gomori trichrome stain show a characteristic pattern, with the eosinophilic mitochondria dumped in shaggy clusters around the periphery of the cell. This appearance has given rise to the term ragged red fibers to describe the histology of Kearns-Sayre syndrome and other mitochondrial disorders. Under the electron microscope, the mitochondria themselves are seen to be abnormal; they are
often enlarged, elongated, or fused, with loss of the usual folding pattern of the internal cristae, and they may contain crystalline inclusion bodies sometimes described as having a so-called railway track or parking lot appearance, in reference to their highly regular, blocklike array. Mitochondria in the myocardium have also been shown to be abnormal in patients with cardiac involvement although the inclusion bodies have typically been more amorphous and unlike the rectilinear crystals noted in skeletal muscle.
often enlarged, elongated, or fused, with loss of the usual folding pattern of the internal cristae, and they may contain crystalline inclusion bodies sometimes described as having a so-called railway track or parking lot appearance, in reference to their highly regular, blocklike array. Mitochondria in the myocardium have also been shown to be abnormal in patients with cardiac involvement although the inclusion bodies have typically been more amorphous and unlike the rectilinear crystals noted in skeletal muscle.
Arrhythmogenic Right Ventricular Dysplasia
Arrythmogeneic right ventricular dysplasia (ARVD) is a primary myocardial disorder characterized by fatty infiltration of the right ventricle, progressive loss of myocytes, inflammatory infiltrate, thinning and dilatation of the ventricular wall, and replacement fibrosis. The right ventricle is affected predominantly; however, both interventricular septal and left ventricular involvement have been reported. Usually manifested by ventricular arrhthymias with left bundle configuration, electrocardiographic abnormalities (including an epsilon wave and sudden-death heart failure) are a less-common feature. The fibrofatty infiltration progresses to yield systolic dysfunction of the right ventricle and, in advanced cases, the left ventricle as well. A negative endomyocardial biopsy does not exclude the diagnosis, and consensus diagnostic criteria include endomyocardial biopsy, echocardiography, electrocardiography, and magnetic resonance imaging (MRI). ARVD is a genetic disorder. Eight loci have been mapped and three genes identified. Autosomal dominant transmission is the most common transmission, but an autosomal recessive has been reported (144,145,146,147,148).
Alcohol-Induced Cardiomyopathy
The major difficulty with this diagnosis is that cardiomy-opathy is a relatively uncommon event and alcohol abuse is a common condition. Clinically and histologically, the overt response to general medical therapy is not significantly different in these patients than in those with idiopathic cardiomyopathy. Alcohol abuse has been identified as a significant risk factor in cardiomyopathy with a frequency of between 20% and 30% (149,150).
It is clear that high blood levels of alcohol produce mildly negative inotropism (151,152). Furthermore, several animal studies have investigated the effects of prolonged alcohol administration. In primates, long-term alcohol administration results in myocytolysis and fibrosis (153). In dogs, cardiac fibrosis is produced, leading mainly to measurable myocardial dysfunction (154,155). In a hamster model, long-term alcohol administration resulted in a reduction in myocardial high-energy phosphates (156).
The specific mechanisms by which alcohol might produce myocardial dysfunction have also been investigated. Cellular events that have been described with long-term alcohol exposure include impaired sarcoplasmic reticular uptake of calcium (157), inhibition of myosin ATPase (158), elevation of intracellular Na+ and water (159), inhibition of the Na+,K+-ATPase (160), and alterations in the incorporation of membrane fatty acid and phospholipids (155,161).
The expression of alcohol-induced cardiomyopathy is obviously quite variable. There is evidence to suggest that excessive alcohol consumption is likely to be present for a minimum of 10 years before the onset of heart failure (162,163,164), that men are more susceptible than women (165), and that concurrent smoking, hypertension, and malnutrition may be contributing factors. Furthermore, there appear to be differences between persons in organ susceptibility to alcohol, as chronic cirrhosis is unlikely to develop in those with cardiomyopathy (166).
An interesting, likely unrelated phenomenon occurred in the 1960s when cobalt was added to beer as a foam-stabilizing agent in a few areas of Europe and Canada. An acute and aggressive form of cardiomyopathy developed in heavy beer drinkers that was attributable to the cobalt (167,168). When this element was no longer used, no further cases of this form of cardiomyopathy were reported.
There is some evidence to suggest that abstaining from alcohol has a beneficial effect on prognosis in alcohol-induced cardiomyopathy. In one study of 64 patients, approximately one-third discontinued excessive alcohol use. This subgroup had a 9% mortality rate during the subsequent 4 years, contrasted with a 57% mortality rate in the remainder who continued drinking (31). A second but smaller study (31 patients) supported these findings (169).
Cocaine and Cardiomyopathy
Long-term cocaine abuse may result in the development of a dilated cardiomyopathy. Termed cocaine-induced cardiomyopathy, cocaine’s direct toxicity to the myocardium may be responsible. These direct effects include provocation of coronary spasm, increased catecholamine release, enhanced platelet aggregation, decreased fibrinolysis, and upregulation of tissue plasminogen activator inhibitors (predisposing the abuser to coronary thrombosis) (170,171,172). Hypersensitivity myocarditis due to cocaine or associated contaminants may be another mechanism (173). Finally, cocaine may induce lethal arrhythmias. Similar to alcoholic cardiomyopathy, abstinence is crucial in therapy and cessation-related reversal of dysfunction has been reported (174).
Treatment with β-blockade was popular in the 1980s; however, reports of cocaine-induced hypertension or myocardial ischemia were thought to be due to use of β-blockers and the resulting unopposed α-receptors. Thus, treatment with β-blockers decreased. While no significant data exist, utilization of labetalol or carvedilol (both of which have α- and β-receptor antagonist activities) may warrant further investigation (175).
Diabetic Cardiomyopathy
Diabetic cardiomyopathy as a unique entity has only recently been recognized. In 1972, Rubler et al. (176) presented data from four patients with Kimmelstiel-Wilson disease and dilated cardiomyopathy in the absence of coronary, valvular, or other cardiomyopathic risk factors. Supportive data have included a retrospective analysis from the Framingham Heart Study demonstrating a 2.4-
fold higher incidence of heart failure in diabetics on insulin (177) and a higher-than-expected incidence of diabetes in a population of patients with idiopathic dilated cardiomyopathy (178). Potential pathogenetic mechanisms for diabetic cardiomyopathy have included increased vascular permeability resulting in increased gly-cosylation of collagen (179), alterations in intracellular Ca2+ kinetics (180), and microvascular damage (181) akin to that of diabetic retinopathy and nephropathy.
fold higher incidence of heart failure in diabetics on insulin (177) and a higher-than-expected incidence of diabetes in a population of patients with idiopathic dilated cardiomyopathy (178). Potential pathogenetic mechanisms for diabetic cardiomyopathy have included increased vascular permeability resulting in increased gly-cosylation of collagen (179), alterations in intracellular Ca2+ kinetics (180), and microvascular damage (181) akin to that of diabetic retinopathy and nephropathy.
The clinical presentation of diabetic cardiomyopathy is not unlike that of idiopathic dilated cardiomyopathy. In a series of 16 diabetics with cardiomyopathy, cardiac volumes, LV filling pressures, and ventricular mass were increased in all patients and cardiac index was reduced in approximately half (178). These findings were confirmed in a small series that also demonstrated a decrease in myocardial compliance (182). Several investigators have demonstrated that diabetics have abnormal cardiac function based on systolic time intervals (183,184,185). Seneviratne noted these abnormalities to be present only in diabetic patients with evidence of other microangiopathy (retinopathy or nephropathy) (184).
Histopathologically, hearts from diabetic patients with dilated cardiomyopathy are similar to those from nondia-betic patients. Some investigators have reported, in addition to the hypertrophy and interstitial fibrosis, an increase in interstitial material positive for periodic acid-Schiff (PAS), consistent with glycoprotein (186). Other investigators have reported hyalin thickening of small vessels in the myocardium (187).
Peripartum Cardiomyopathy
The syndrome of unexplained heart failure in women post-partum was detailed in the mid-1930s by a number of investigators (188,189) but had been suggested even earlier. The disease was initially described as occurring primarily in the postpartum period, but it is now clear that it can present as early as the end of the middle of the third trimester or as late as several months after delivery. Most commonly, however, signs and symptoms of heart failure occur postpartum (190).
The etiology of peripartum cardiomyopathy is unknown, although several factors have been associated with development of the disease. As the disease appears to be more common in less-affluent populations, nutritional deficiencies during pregnancy have been postulated (191,192). Other suggested associations have included toxemia of pregnancy (192), immunologic factors such as the development of antiheart antibodies (193), genetic susceptibility (194,195), and drug-induced hypersensitivity (196).
Although the general clinical signs, symptoms, and histology (except for patients with myocarditis) are no different from those of patients with idiopathic dilated cardiomyopathy, prior series suggested certain associated factors, including race (with a much higher incidence in African-Americans), advanced maternal age, multiparity, and twin pregnancies (190,197). A more recent series of 123 patients suggested otherwise. While patients were somewhat older (mean age of 31), most (67%) were white, gestational hypertension was present in almost half of the patients, and tocolytic therapy was used in 19%; only 13% were twin pregnancies (198).
This entity is undoubtedly distinct from other forms of dilated cardiomyopathy, as evidenced by three well-documented phenomena. First, as previously stated, the onset of disease in most cases is close to the postpartum period (197). Second, relatively speaking, the prognosis of peripartum cardiomyopathy is excellent compared with the prognosis in other forms of dilated cardiomyopathy. With current medical therapy based on the most recent series of patients, 54% of patients had normalization of their ejection fraction (198). Third, the disease appears to recur during subsequent pregnancies in a portion of patients, even those who have completely recovered from their first episode (194,199).
Anthracycline-Induced Cardiomyopathy
This form of toxin-induced cardiomyopathy may affect anywhere from 5% to 20% of patients receiving the agent, depending on a variety of risk factors and total dose. This is a unique form of cardiomyopathy that has provided several insights into toxin-induced cardiac disease and global cardiac function for the following reasons: First, as a toxin, anthracycline appears to affect certain cardiac cells but not others, according to electron microscopic studies (200,201). The number of affected cells appears to be related to total dose. This is presumably a different phenomenon from that noted in other forms of dilated cardiomyopathy, in which the insult or defect appears to affect cardiac tissue more diffusely. Second, there appears to be a threshold effect, in which gross cardiac function remains normal until a certain percentage of cells has been affected; cardiac function can then deteriorate precipitously (202). This would suggest a partial redundancy in the number of contractile units (myocytes) in the heart, similar to that in the functioning units of other organs (e.g., glomeruli).
Several hypotheses have been proposed to explain the cardiotoxic effect of anthracyclines. Oxygen free radicals, lipid peroxidation, altered arachidonic acid metabolism, a direct interaction with the calcium release from sarcoplas-mic reticulum, and metabolite action on the calcium pump from sarcoplasmic reticulum have all been implicated in the cardiotoxic effects (203).
The clinical presentation of cardimyopathy secondary to anthracycline toxicity is not dissimilar to that of dilated cardiomyopathy of other etiologies. It has been reported as early as a week following the last dose of doxorubicin but can present even years after chemotherapy (204). It has been suggested that the shorter the latency period, the greater the severity and the poorer the ultimate prognosis (204). Most studies have suggested that cardiac toxicity is uncommon in cumulative doses of less than 450 mg/m2 in the absence of other risk factors (204). When risk factors are present (which include advancing age, mediastinal radiation, concomitant cardiotoxic chemotherapy, underlying cardiac disease, and large bolus therapy) cardiac toxicity has been noted at substantially lower total doses (204,205,206,207,208).
Although a variety of cardiovascular studies have been advocated to follow patients for cardiac toxicity, the most sensitive appears to be endomyocardial biopsy (202). This can in part be attributed to the threshold phenomenon previously described. Histopathologic findings include disruption of myofibrils and dissolution of myofilaments, extensive
vacuolization secondary to swelling of the sarcoplasmic reticulum, mitochondrial pleomorphism, and dissociation of the tight junctions (200,201). These findings are graded according to the number of cells affected and severity of the changes. Most noninvasive studies, such as echocardiography, radionuclide angiography, and systolic time intervals, rely on systolic cardiac function, which may not fall despite significant histological evidence of toxicity (209). It is conceivable that diastolic function might provide more sensitive data for following these patients.
vacuolization secondary to swelling of the sarcoplasmic reticulum, mitochondrial pleomorphism, and dissociation of the tight junctions (200,201). These findings are graded according to the number of cells affected and severity of the changes. Most noninvasive studies, such as echocardiography, radionuclide angiography, and systolic time intervals, rely on systolic cardiac function, which may not fall despite significant histological evidence of toxicity (209). It is conceivable that diastolic function might provide more sensitive data for following these patients.
While anthracycline cardiac toxicity has been reduced, first, by more careful monitoring of toxicity and a better understanding of risk factors, patients with impaired LV dysfunction should probably receive anthracyclines only as a last resort. Even with normal LV function, once the total dose of doxorubicin exceeds 450 mg/m2 (or less if risk factors are present), histological monitoring by endomyocardial biopsy is warranted before additional drug is administered. A highgrade biopsy specimen should preclude additional anthracycline exposure irrespective of LV systolic function. Second, it appears that toxicity can be reduced by reducing peak levels of the drug, so that protocols have been changed from monthly large infusions to weekly smaller infusions and even daily continuous intravenous administration. It has been observed that larger total doses can be achieved, efficacy is not impaired, and toxicity is lessened (209).
Trastuzumab (Herceptin)-lnduced Cardiomyopathy
Trastuzumab is a relatively new monoclonal antibody directed against the HER2/erbB2 recepter that is overexpressed on many breast carcinomas (CA). It is being used more commonly in both the adjuvant as well as treatment settings for breast CA and, not infrequently, in combination with doxorubicin and other anthracyclines. In a subset of individuals, trastuzumab induces a resersible cardiomyopathy. The mechanism of cardiac toxicity is unknown, but the cardiomyocyte does express low levels of HER2/erbB2. In vitro data suggest that despite these low levels, the cardiac receptor appears to be uniquely susceptible to trastuzumab in the subset of individuals where there is an interaction between trastuzumab and the cardiac HER2/erbB2 receptor (210). In most patients the cardiac dysfunction is reversible with discontinuation of therapy (210).
Left Ventricular Dysfunction Associated with Inflammation
A number of infectious agents and two major inflammatory conditions have been associated with left ventricular dysfunction. These are outlined in Table 18-2.
Viral Myocarditis
Viral myocarditis follows a pattern. The virus is usually introduced through the respiratory or gastrointestinal tract. In the first stage (the first 3 days), viremia results in acute myocarditis by way of myocardial invasion, viral replication, nuclear transmission, release to the cell surface, and subsequent infection of other cells. The first response to myocarditis is a nonspecific immune response. Nuclear factor kappa B (NF-KB), an intracellular transcription factor
which produces cytokines, nitric oxide, and intracellular adhesion molecules, is virus-activated. The inflammatory response to cardiac viral infection involves all three substances resulting from NF-KB. Lung or gut infection results in cytokine expression, which may lead to a cardiac immune response. The invasion of myocytes results in cell necrosis and macrophage activation. Macrophage activation results in cytokine expression, including tumor necrosis factor-alpha (TNF-α), interleukins, and interferon (211,212,213,214,215,216).
which produces cytokines, nitric oxide, and intracellular adhesion molecules, is virus-activated. The inflammatory response to cardiac viral infection involves all three substances resulting from NF-KB. Lung or gut infection results in cytokine expression, which may lead to a cardiac immune response. The invasion of myocytes results in cell necrosis and macrophage activation. Macrophage activation results in cytokine expression, including tumor necrosis factor-alpha (TNF-α), interleukins, and interferon (211,212,213,214,215,216).
Table 18-2 Principal Etiological Agents Associated with Myocardits | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The second stage (subacute myocarditis, days 3 to 14) is an immunologic response and consists of the infiltration of mononuclear cells, B lymphocytes, and cytotoxic T cells. Natural killer cells limit viral replication and release perforin, forming lesions on the infected cells’ membrane surfaces, thus sparing noninfected cells from injury. Macrophages and dendritic cells ingest viral agents and are responsible for the initial cell-mediated response to viral myocarditis by releasing proteases, cytokines, perforin, and interleukins. Nitric oxide (NO) is increased in macrophages in the setting of myocarditis. NO plays a role in this phase, which may be beneficial or detrimental by enhancing or suppressing myocardial function. In animal models, NO may contribute to the progression of myocyte damage (217,218,219,220,221).
The third stage (chronic myocarditis, days 14 to 90) is marked by fibrosis, cardiac dilatation, and heart failure. Possible mechanisms of progression to dilated cardiomyopathy include viral persistence, apoptosis, coronary microvascular spasm, and autoimmunity. Persistence of virus (although the virus cannot multiply, it may be housed in the skeletal muscle) coupled with an activated immune system is the first possible mechanism. Apoptosis (programmed cell death) is a second possible mechanism of progression to dilated cardiomyopathy and may be triggered by certain viruses. Coronary microvascular spasm results in myocyte necrosis. Finally, a third possible mechanism is the role of autoimmunity in this progression by way of antibody to viral proteins crossreacting with normal heart architecture (222,223,224,225).
Clinical manifestations are variable, ranging from the asymptomatic patient to cardiogenic shock. Cardiac involvement occurs days to weeks subsequent to a viral illness. The clinical presentation of cardiac inflammation consists of symptoms due to heart failure, arrhythmia, and embolic events. Myocarditis-associated syncope may be due to ventricular arrhythmias or atrioventricular (AV) block. The AV block is usually transient Sudden cardiac death due to myocarditis has been reported. Patients note symptoms of a preceding viral illness followed by myalgias, fever, palpitations, dyspnea, and chest pain. Chest pain may be pleuritic due to simultaneous pericardial inflammation, typical of myocardial ischemia, or atypical. The cardiac exam demonstrates tachycardia, gallops, murmurs of AV valve regurgitation, or possibly a pericardial friction rub. A presentation identical to acute myocardial infarction may occur including chest pain, ST elevation, elevation of the MB fraction of focal or diffuse wall motion abnormalities, a normal coronary angiogram, and biopsy-proven myocarditis. Both viral arteritis of the coronary arteries and vasospasm have been reported. Ventricular microaneurysms due to myocarditis are diagnosed by LV biopsy in the setting of preserved systolic function and nonsustained ventricular tachycardia (226,227,228,229,230,231,232,233,234,235).
Laboratory findings are nondiagnostic. The white blood cell count is usually elevated, and eosinophilia and elevation of the erythrocyte sedimentation rate are sometimes noted. Cardiac-specific troponin I is elevated in up to one-third of patients, while CK-MB is increased in 10% of patients. The electrocardiogram demonstrates ST-T wave changes, supraventricular or ventricular ectopy, atrial fibrillation, conduction abnormalities, low voltage, and prolongation of the QTc interval. Rarely, an acute infarct pattern is seen. The most frequent electrocardiogram finding is sinus tachycardia. Cytokine elevation, including IL-6, TNG, and IgG3 may be seen in patients with progressive ventricular dysfunction (236,237,238).
Echocardiographic findings include ventricular thrombi, increased wall thickness, and systolic dysfunction with possible segmental wall motion abnormalities. MRI may identify myocarditis-induced tissue alterations (239,240,241,242,243).
Endomyocardial biopsy can confirm the diagnosis if the clinical diagnosis is confusing. The diagnosis of active myocarditis requires an inflammatory infiltrate of the myocardium with necrosis and/or degeneration of adjacent myocytes not typical of the ischemic damage associated with coronary artery disease (244).
Supportive therapy for heart failure in general is utilized in the setting of myocarditis. Patients should limit their physical activity. Diuretics are used for relief of congestive signs and symptoms. Steroids prolong viremia in rats. Supportive care with angiotensin-converting enzyme (ACE) inhibitors should be employed. Digoxin, which increases the expression of cytokines in animal studies, is used both cautiously and at a low dose due to concern for precipitating vascular spasm. Data regarding the use of calcium channel antagonists are lacking (245,246,247).
Immunosuppressive therapy has been evaluated in studies with discouraging results. Intravenous immunoglobulin was not shown to be beneficial in a trial of 62 patients with heart failure, only 16 of whom had myocarditis per the Dallas criteria (248).
The Myocarditis Treatment Trial evaluated the response to conventional therapy versus prednisone in combination with either cyclosporine or azathioprine in patients with biopsies compatable with myocarditis (nonviral) per the Dallas criteria. No difference was found in any group in terms of ejection fraction increase, mortality, or disease attenuation. Parillo reported the effect of prednisone in 102 patients with established dilated cardiomyopathy (not viral myocariditis), finding no lasting improvement in ejection fraction after 9 months. Routine use of immunosuppressive therapy cannot be recommended. Antiviral therapy is currently being investigated (249,250).
Rickettsial Myocarditis
Myocarditis due to rickettsial disease is common, although usually subclinical. ST-T wave changes are frequently noted. Vasculitis and perivascular inflammation are pathologic features, and peripheral vascular abnormalities (with or without accompanying myocarditis) are responsible for circulatory collapse. Rickettsial infections responsible for myocarditis include Q fever (Rickettsia burnettii), Rocky Mountain spotted fever (R. rickettsii), and scrub typhus (R. tsutsugamushi) (251,252,253,254,255).
Bacterial Myocarditis
Bacterial myocarditis is rare and has been associated with a number of gram-positive and gram-negative organisms. These include but are not limited to organisms responsible for brucellosis, clostridial disease, diptheria, Legionnaire disease, mycoplasma, psittacosis, Salmonella, Streptococcus, and tuberculosis (256,257,258,259,260,261,262,263).
Spirochetal Infections
Spirochetal infections causing myocarditis include syphilis, relapsing fever, and leptospirosis. Leptospirosis, a cause of fatal myocarditis, occurs with ST-T wave changes noted on the surface ECG in half of all cases.
Lyme disease is caused by Borrelia burgdorferi, a spirochete introduced by a tick bite. Erythema chronicum migrans, the characteristic rash of Lyme disease, first appears, with subsequent neurological or cardiac manifestations appearing weeks to months later. Cardiac involvement is seen in approximately in 10% of cases and presents as variable degrees of AV block, ventricular tachycardia, myopericarditis, bundle-branch block, LV dysfunction, transient ST-T wave changes, and syncope due to complete heart block. Endomyocardial biopsy may demonstrate active myocarditis and (rarely) spirochetes. Immune-mediated mechanisms may be involved. The prognosis is good and most patients recover completely. Treatment includes hospitalizing those patients with second- or third-degree AV block and placement of a temporary pacemaker, which may be necessary for a week or longer. While the efficacy of antibiotics has not been established, both intravenous (ceftriaxone or penicillin G) and oral agents have been used. Corticosteroid therapy after treatment with tetracycline may be helpful in treating carditis, but treatment with steroids to ameliorate heart block is controversial (264,265,266,267,268,269,270).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


