Fig. 10.1
Organization of the autonomic nervous system demonstrating the key interactions involving the brain, heart, and kidney. SA sino-atrial node (Reproduced with permission from Singh et al. [159])
10.2 Measurements of Autonomic Nervous System Activity
One important challenge to the understanding of the bidirectional autonomic interactions between the heart and the kidney is the ability to quantify individual regional SNA activity. For this purpose, sympathetic nerve recording techniques and radiotracer-derived measurements of norepinephrine (NE) spillover into the plasma from individual organs have been used. The limitations of each technique have led to the recommendation that they be used together [7]. Microneurography provides instantaneous multiunit or single-fiber recordings of electrical transmission in sympathetic nerves, but assessment may be skewed by interpreter’s bias [8, 9].
The NE spillover method provides objective information on the release of this neurotransmitter from internal organs where microneurography is not feasible [10–12]. During infusion of titrated NE at a constant rate, output of endogenous NE from a given organ (NE “spillover”) can be measured by isotope dilution according to the formula:
![$$ \mathrm{Regional}\kern0.24em \mathrm{norepinephrine}\kern0.24em \mathrm{spillover}=\left[\left({C}_V-{C}_A\right)+{C}_AE\right]\;PF $$](/wp-content/uploads/2016/09/A329012_1_En_10_Chapter_Equa.gif) where C V and C A are the plasma concentrations of NE in the organ’s venous and arterial plasma, E is the fractional extraction of titrated NE while the blood is flowing through the organ, and PF is the organ plasma flow [7].
where C V and C A are the plasma concentrations of NE in the organ’s venous and arterial plasma, E is the fractional extraction of titrated NE while the blood is flowing through the organ, and PF is the organ plasma flow [7].
Computer analysis of heart rate variability (HRV) predominantly reflects selective autonomic control of the heart. Vagal and sympathetic cardiac influences operate on the heart rate in different frequency bands. While vagal regulation has a relatively high cutoff frequency, modulating heart rate both at low and high frequencies (up to 1.0 Hz), sympathetic cardiac control operates only at <0.15 Hz [13–15]. Blood pressure variability is the result of complex interactions between cardiac and vascular neural regulation, mechanical influences of respiration, humoral and endothelial factors, large artery compliance, and genetic influence. Nevertheless, time or frequency domain analysis of either blood pressure or HRV can provide valuable information on autonomic cardiovascular regulation. While often lacking specificity, these measurements can be obtained in clinical practice and are not subject to interpreter’s bias [16]. The ability of HRV and blood pressure fluctuations to reflect autonomic control of the cardiovascular system is improved by use of multivariate models for its assessment. The simplest ones consider the relationship between spontaneous fluctuations in blood pressure and heart rate, either in the time or frequency domain to assess baroreceptor sensitivity (BRS) and its modulation in daily life [17–20]. While spontaneous variations in blood pressure and heart rate clearly depend on autonomic mechanisms, caution is needed in considering them a quantitative measurement of efferent SNA to the heart and vasculature. In fact, in a variety of clinical situations, including HF, low-frequency heart rate spectral power has little or no relation to rates of NE spillover from the heart or sympathetic nerve firing measured by microneurography. Indeed, in HF, low-frequency heart rate spectral power is reduced, but cardiac NE spillover is markedly increased [21].
Cardiac SNA can also be noninvasively assessed by the use of 123I-metaiodobenzylguanidine (MIBG), an analogue of NE, using semiquantitative analyses, namely, early heart-to-mediastinum ratio, late heart-to-mediastinum ratio, and myocardial washout [22]. Data from prospective studies and meta-analyses have shown that patients with decreased late heart-to-mediastinum ratio or increased myocardial 123I-MIBG washout have a worse prognosis than those patients with normal semiquantitative myocardial MIBG parameters [23]. Furthermore, 123I-MIBG has been found to independently predict sudden cardiac death regardless of left ventricular ejection fraction (LVEF) [24]. In addition, the ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) trial demonstrated that 123I-MIBG cardiac imaging provides additional independent prognostic information for risk-stratifying HF patients on top of commonly used markers such as LVEF and B-type natriuretic peptide [25].
Thus, the clinical relevance of information on autonomic cardiac control is supported by the evidence that increased SNA is associated with increased mortality in myocardial infarction and HF patients, and with an increased risk of sudden arrhythmic death.
10.3 Sympathetic Innervation of the Kidney
The kidney is abundantly innervated with both efferent adrenergic and somatic afferent neurons [26, 27] (Fig. 10.2). The efferent neurons terminate at multiple sites within the nephron and independently influence tubular sodium reabsorption, renin secretion, and renal blood flow (RBF). Sodium reabsorption is enhanced at very low stimulation frequencies; higher stimulating frequencies increase renin secretion and lower RBF [28–30]. Thus, under conditions of mild sympathetic activation, sodium reabsorption increases and consequently plasma volume expands. With more intense sympathetic activation, sodium reabsorption is further augmented by the effects of angiotensin II (A II) and aldosterone and vasoconstriction occurs due to the combined vascular effects of NE and A II. Thus, increased efferent renal SNA produces simultaneously an increase in arterial pressure and blood volume and a decrease in RBF. When these compensatory responses to hypotension or hypovolemia become persistent and disproportionate to the cardiovascular abnormalities which initially trigger them, they become maladaptive and directly contribute to the progression of HF. These responses are particularly influential in the CRS where persistent vascular congestion and worsening renal function magnify the mutually detrimental effects of heart and kidney [31]. It should be noted that in addition to the effects of renal sympathetic efferent activation, somatic afferent nerves originating in the kidney act directly on the neural cardiovascular control centers in the midbrain [27]. The activity of these afferent nerves is stimulated by various factors including ischemia and adenosine release, both of which are the result of intense vasoconstriction. A direct neurological connection between the kidney and hypothalamus has been demonstrated in partially nephrectomized rats. In patients with end-stage renal disease (ESRD) and in renal transplant recipients, removal of the native kidney is associated with attenuated muscle SNA [32–35]. Because A II can directly stimulate central sympathetic drive, secretion of renin by the macula densa cells is another mechanism by which the kidney contributes to activation of SNA and of the renin-angiotensin-aldosterone system (RAAS) [36]. Increases in activation of either the afferent or the efferent loop of this “sympathorenal axis” may lead to a self-perpetuating cycle and sustained generalized SNA. However, it should be pointed out that many other reflexes and humoral substances, including natriuretic peptides (NP), can modify sympathetic tone, so that any contribution of the renal sympathetic afferent nerves to this self-perpetuating cycle can be modified by changes in activity of these other controllers [22].
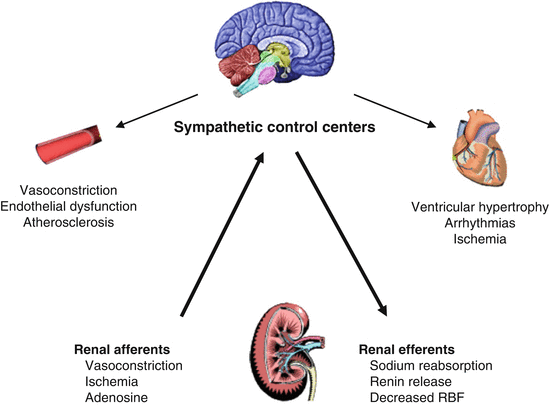
Fig. 10.2
Afferent sympathetic pathways travel from the kidney to the control centers for neuromodulation in the midbrain. Activation of these pathways increases global sympathetic traffic, which may adversely affect vascular tone and integrity, as well as lead to inappropriate myocardial hypertrophy, myocardial cell damage, and arrhythmias. Increased renal sympathetic signaling stimulates sodium retention, volume expansion, and renal vasoconstriction. The consequences of increased renal sympathetic efferent traffic may also lead to an increase in afferent traffic, thereby creating a positive feedback loop with many deleterious vascular, myocardial, and renal consequences. RBF renal blood flow (Reproduced with permission from Goldsmith et al. [39])
10.4 The Sympathorenal Axis in Heart Failure
Increased plasma NE, muscle SNA, and total body, cardiac, and renal NE spillovers have been documented in patients with congestive HF [12, 37]. For more than three decades, plasma NE has been known to be a strong predictor of outcomes in HF patients [38]. The success of beta-blockers in decreasing mortality in HF patients convincingly supports the notion that excessive SNA directly contributes to HF progression [39].
However, the use in HF patients of moxonidine, an inhibitor of presynaptic NE release, was associated with increased mortality presumably due to hypotension caused by a precipitous fall in plasma NE levels [40]. Data are lacking on whether a more gradual reduction in NE levels would have produced different results in HF patients.
There is little doubt that renal NE spillover is, together with cardiac NE spillover, a major contributor to the total excess of sympathetic drive in HF patients [12]. Indeed, it has recently been shown that renal SNA, as measured by renal NE spillover, was highly predictive of outcomes despite concomitant therapy with anti-neurohormonal drugs [39]. In addition in an experimental myocardial infarction model, renal sympathetic denervation was associated with improved outcomes [41]. The enhanced efferent sympathetic signaling to the kidney seen in HF presumably has the same effects it has in hypertension (enhanced sodium retention, decreased RBF and activation of the RAAS). These effects are even more harmful in HF because volume expansion and increased arterial pressure will aggravate myocardial loading conditions and, together with the direct actions of NE, A II, and aldosterone, worsen myocardial remodeling. The SNA-related sodium avidity and renal hemodynamic abnormalities may be especially deleterious in the CRS, because persistent congestion may itself contribute to further deterioration of renal function [31]. Kidney dysfunction often occurs during intensive treatment with loop diuretics. This event is not surprisingly because loop diuretics are known to further stimulate SNA either directly or through activation of the RAAS [42]. Augmentation of afferent signaling from the kidney may then contribute to perpetuate the global sympathetic overdrive in HF, completing the sympathorenal loop [39] (Fig. 10.3).
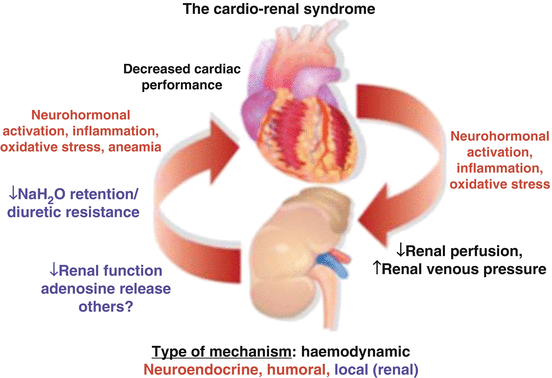
Fig. 10.3
Cardiorenal interactions in heart failure and kidney disease. Most of the mechanisms may be activated by each of the two conditions and are able to affect both cardiac and renal function. The mechanisms involved in the pathologic interactions between the heart and the kidney include hemodynamic abnormalities, neurohormonal activation, inflammation, and local intrarenal events (Reproduced with permission from Metra et al. [213])
10.5 Consequences of Congestion and Neurohormonal Activation in the Kidney and in Other Regional Circulatory System in the Abdomen
Many critically important changes occur in the kidney in the setting of cardiac dysfunction and neurohormonal activation (Fig. 10.3). When RBF decreases as a result of decreased cardiac output (CO), neurohormonally induced efferent arteriolar vasoconstriction, or increased central venous pressure (CVP), the kidney strives to maintain glomerular filtration rate (GFR) by increasing filtration fraction (FF) [43]. In normal conditions, FF is approximately 20–25 %, increases above this value in HF, and can rise above 50 % when congestion is complicated by increased intra-abdominal pressure. As explained below, increased FF in itself augments sodium reabsorption, an event which is magnified by increased SNA and RAAS activation. Different transporters mediate active transfer of sodium across the luminal side of proximal tubular cells. However, because the proximal tubules have a highly permeable epithelium, sodium can easily return to the lumen so that net sodium reabsorption is governed by passive Starling forces between the peritubular capillaries and renal interstitium. In congestive HF, because of an increased FF, the oncotic pressure in the peritubular capillaries (πPC) is higher, which stimulates sodium and water reabsorption into the vasculature. Because the kidney is an encapsulated organ, when congestion is present, the interstitial fluid hydrostatic pressure (PIF) and the peritubular capillaries hydrostatic pressure (PPC) are both increased, whereas the interstitial fluid oncotic pressure (πIF) drops because of increased lymph flow, which removes interstitial proteins. This also favors net sodium and water reabsorption into the vasculature [44–51]. Abnormally high-sodium reabsorption in the proximal tubule has profound consequences on the rest of the nephron. Under normal circumstances, the macula densa senses increased sodium chloride delivery because active chloride transport requires ATP, which is ultimately converted to adenosine. This substance, which is released from cells of the macula densa, has a paracrine vasoconstrictive effect on the afferent arteriole. This effect, known as tubuloglomerular feedback (TGF), protects the glomerulus from hyperfiltration injury. In congestive HF, due to increased sodium chloride reabsorption in the proximal tubule, chloride delivery to the macula densa is reduced and intracellular chloride levels are low. This stimulates NOS I and COX-2 activation and release of NO and PGE2. Both NO and PGE2 stimulate the granulosa cells of the afferent arteriole to secrete renin which activates angiotensin II, thus perpetuating a vicious cycle of neurohormonal activation and worsening congestion. It is also important to consider that loop diuretics, which are used in large numbers of ambulatory and in the majority of hospitalized HF patients, inhibit the Na+/K+/2Cl− co-transporter in the thick portion of the ascending loop of Henle, further reducing macula densa uptake of sodium chloride and escalating neurohormonal activation [43]. The distal convoluted tubules and collecting ducts reabsorb ≤10 % of the total amount of sodium filtered by the glomerulus. In contrast to the part of the nephron proximal to the macula densa, where net fractional sodium reabsorption is kept relatively constant under normal circumstances, distal fractional sodium reabsorption rates are highly variable depending on tubular flow rate, and levels of aldosterone and arginine vasopressin [52–54]. Therefore, it is the distal nephron which determines the urinary sodium concentration and osmolality. However, a prerequisite for the ability of the distal nephron to maintain a neutral sodium balance is adequate delivery of sodium. In congestive HF, because of increased fractional reabsorption in the proximal tubules and often decreased GFR in individual nephrons, tubular flow might be low in the distal part of the nephron despite significant systemic fluid excess. In addition, the increased levels of aldosterone and arginine vasopressin further stimulate reabsorption of the remaining tubular fluid. It is the decreased distal tubular flow which causes aldosterone breakthrough, which leads to secondary hyperaldosteronism despite therapy with adequate doses of RAAS inhibitors [55, 56]. Furthermore, prolonged exposure to loop diuretics produces adaptive hypertrophy of distal tubular cells, which increases local sodium reabsorption and aldosterone secretion. Indeed, experimental data shows that distal tubular cells adaptation to loop diuretics can be significantly attenuated by administration of aldosterone antagonists or thiazide diuretics [55, 57].
The escalating congestion resulting from the cardiorenal interactions outlined above has profound implications for all abdominal vascular systems, including the splanchnic, intestinal, hepatic, and splenic circulations [58]. In the splanchnic microcirculation, net filtration rate is determined by Starling forces, (PC -PIF) – (πC- πIF), which favor filtration throughout the entire length of the capillary bed. When capillary hydrostatic pressure increases as a result of congestion, filtration pressure is even higher [58]. Because the interstitium has low compliance, the excess filtrated fluid is drained directly into lymphatic capillaries, so that there is only a slight increase in interstitial fluid volume. Splanchnic lymphatic flow can increase as much as 20 times its normal value [59]. Because increased lymphatic flow removes interstitial proteins, the drop in interstitial oncotic pressure reduces filtration and the accumulation of fluid in the interstitium. Once lymphatic flow cannot increase further and interstitial compliance increases, interstitial fluid begins to accumulate [60]. When lymph flow can no longer adequately remove interstitial proteins, protein-rich edema accumulates to the point of compressing lymphatic vessels which further impairs lymph flow.
The intestinal microcirculation is characterized by a countercurrent system that enables extensive exchange of oxygen (O2) between arterioles and venules. This O2 “short circuit” creates a gradient with the lowest partial O2 pressure at the villus tip [61–63]. During congestive HF, low perfusion, venous congestion, and sympathetically mediated arteriolar vasoconstriction in the splanchnic microcirculation stimulate O2 exchange between arterioles and venules, exaggerating the O2 gradient between the villus base and tip. This causes villus tip ischemia which is responsible for epithelial cells dysfunction and loss of intestinal barrier function. As a result, lipopolysaccharide or endotoxin, produced by gram-negative bacteria residing in the gut lumen, enters the systemic circulation and contributes to escalate the HF inflammatory milieu [61–63].
In the liver, hepatocytes continuously produce adenosine from the breakdown of ATP. Adenosine accumulates in the perisinusoidal space, which is drained by the lymphatic system. When portal blood flow is reduced because of α receptor–mediated vasoconstriction, lymph flow decreases and intrahepatic adenosine concentration increases. Adenosine then stimulates hepatic afferent nerves, which have synaptic connections with renal efferent sympathetic nerves. These events intensify renal vasoconstriction and sodium retention [58].
The splenic sinusoids are freely permeable to plasma proteins. As a result, their colloid osmotic pressure is the same as that of the surrounding lymphatic matrix. Therefore, fluid transport between the two spaces is dictated by differences in hydrostatic pressure. Transient congestion of the splanchnic venous system results in increased hydrostatic pressure inside the splenic sinusoids so that more fluid is transferred to the lymphatic matrix and buffered inside the lymphatic reservoirs of the spleen [64]. In congestive HF, increased cardiac filling pressures increase the production of atrial natriuretic peptide (ANP) which produces splenic arterial vasodilatation and venous vasoconstriction. These hemodynamic changes promote the shift of fluid into the perivascular third space of the spleen. Storage of large amounts of fluid in the spleen may lead to perceived central hypovolemia which further stimulates neurohormonal activity and perpetuates the vicious circle of congestion-driven SNA and RAAS enhancement. Moreover, when the splenic lymphatic circulation becomes overloaded, additional accumulation of interstitial edema occurs [58].
10.6 Autonomic Crosstalk in the Different Types of Cardiorenal Syndrome
According to a widely accepted definition, the cardiorenal syndrome is a pathophysiologic disorder of the heart and kidneys whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction in the other organ. On the basis of this definition, five types of the cardiorenal syndrome have been identified and each has unique aspects of autonomic crosstalk between the heart and the kidney [65].
10.6.1 Cardiorenal Syndrome Type 1
This type of CRS is defined as abrupt worsening of cardiac function, such as it occurs with acute cardiogenic shock or ADHF, leading to acute kidney injury (AKI). Hemodynamic abnormalities play a crucial role in the pathogenesis of the CRS type 1 and trigger decreased renal arterial flow, renal oxygen consumption, and GFR and increased renal vascular resistance [66].
Different ADHF hemodynamic profiles have been identified on the basis of individual patients’ adequacy of perfusion, assessed by measurement of CO, and extent of increase in cardiac filling pressures [67]. Since patients’ clinical characteristics, treatment, and outcomes vary for different hemodynamic profiles, the pathophysiology of CRS type 1 may differ according to the hemodynamic milieu in which it occurs [67, 68]. The occurrence of AKI during ADHF is not restricted to an individual hemodynamic profile and may in fact be related to shifts in hemodynamic conditions when ADHF worsens or in response to treatment.
When CRS type 1 develops in patients with significant reductions in CO, with or without an increase in cardiac filling pressures, it is highly likely to be associated with a reduction in RBF. In ADHF, the relationship between CO, RBF, and intrarenal blood flow distribution remains unclear. However, it is plausible that activation of SNA and RAAS resulting from a significant reduction in intravascular volume will cause renal afferent (and, to a lesser extent, efferent) arteriolar vasoconstriction, leading to a decrease in RBF and renal perfusion pressure. If a low CO is associated with systemic arterial hypotension, renal perfusion pressure may decrease despite a relatively normal renal venous pressure, because renal autoregulation may be unable to compensate for the low blood pressure if the intravascular fluid volume is reduced. The finding of severely decreased RBF and GFR in the setting of reduced CO can therefore indicate that renal autoregulation is impaired [65].
An elevated CVP, which is readily transmitted to the renal vein, directly influences renal perfusion pressure [68–71]. In addition, because the kidney is an encapsulated organ, high renal venous pressure increases renal interstitial hydrostatic pressure. If this exceeds tubular hydrostatic pressure, the tubules collapse. Consequently, increasing intratubular pressure opposes filtration and therefore decreases GFR [65]. This mechanism is supported by experimental data showing a linear decrease in GFR upon increases in renal venous pressure, especially during volume expansion [72]. The response of renal autoregulation to increased renal venous pressure is unknown. However, it has been proposed that higher intrarenal levels of angiotensin II and SNA can indirectly influence arteriolar tone [72, 73].
It is more difficult to explain how the CRS type 1 occurs in ADHF patients with relatively preserved CO. If RBF is sufficiently reduced to be associated with a decreased GFR, it follows that in this situation, the drop in RBF is disproportionate to the decline in CO. This can occur with uni- or bilateral renal artery stenosis, which is estimated to occur in up to 40 % of patients with coexisting HF and CKD [74]. Other factors that may contribute to the development of the CRS type 1 in ADHF patients with preserved CO include (a) chronic use of RAAS inhibitors that impair the renal autoregulatory response to reductions in intravascular volume, (b) use of nonsteroidal anti-inflammatory drugs (NSAID) which may block TGF that would normally produce afferent arteriolar vasodilatation in response to a decreased intravascular fluid volume, and (c) preexisting arterial hypertension which may be associated with a reduction in functional nephrons [43, 65, 75].
It is also important to note that in patients with relatively preserved CO, the relative impact of increased CVP on renal perfusion pressure may not be as high as in patients with intravascular volume depletion. However, according to the mechanisms described earlier, an elevated CVP can still reduce GFR due to increased renal interstitial pressure and neurohormonal activation within the kidney and other regional circulations [67–71].
Biomarker evidence of neurohormonal activation includes the elevation in the levels of natriuretic peptides, mid-regional pro-adrenomedullin, and copeptin [75]. Indeed, ADHF patients with baseline elevation of natriuretic peptide levels and concomitant increase in cardiac filling pressures are at the highest risk for the development of the CRS type 1 [76].
10.6.2 Cardiorenal Syndrome Type 2
This type of CRS is characterized by chronic abnormalities in cardiac function leading to kidney injury or dysfunction. As such, the temporal relationship between the heart and kidney disease is an important aspect of the definition. While observational data clearly show that chronic heart and kidney disease commonly coexist, most studies lack information on whether cardiac dysfunction truly preceded renal abnormalities [77–80]. Thus, the mere coexistence of cardiovascular disease and CKD is not sufficient to make a diagnosis of true CRS type 2 which requires evidence that HF is the underlying cause of the onset and progression of CKD. One clear example is that of an acute myocardial infarction resulting in chronic LV dysfunction followed by the onset of renal function impairment or progression of preexisting CKD. The predominant mechanisms leading from cardiac to renal dysfunction include neurohormonal activation, chronic renal hypoperfusion and venous congestion, inflammation, and oxidative stress. In addition, recurrent ADHF hospitalizations may contribute to the onset and progression of renal impairment [81] (Fig. 10.4). In patients with HF, the frequency of HF admissions has been shown to be independently associated with development of CKD most likely as a result of recurrent episodes of AKI caused both by the hemodynamic abnormalities and the treatment of ADHF [82]. In fact, animal models and epidemiologic studies have shown that repeated episodes of AKI lead to the development and progression of CKD [83].
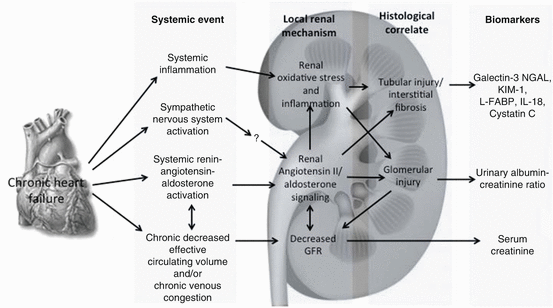
Fig. 10.4
Predominant pathophysiologic mechanisms of CRS2 in stable chronic HF. NGAL neutrophil gelatinase-associated lipocalin, KIM-1 kidney injury molecule-1, L-FABP L-Fatty acid binding protein, IL-18 interleukin-18, GFR glomerular filtration rate (Reproduced with permission from ADQI. Reproduced from Acute Dialysis Quality Initiative 10, under the terms of the Creative Commons Attribution License. Available at: www.ADQI.org. Accessed 2 July 2015)
As described earlier in the chapter, experimental models of chronic HF have shown that neurohormonally induced efferent arteriolar vasoconstriction in response to reduced glomerular plasma flow increases FF to preserve GFR. When these hemodynamic abnormalities persist for extended time periods (3–6 months in the rat model), glomerular pathologic changes ensue, as evidenced by albuminuria, podocyte injury, and focal glomerulosclerosis [43]. Eventually, when increased FF becomes inadequate to preserve filtration in individual glomeruli, GFR begins to decline. Importantly, many of these changes appear to be related to systemic and local renal increases in SNA and RAAS activation [43].
One of the principal roles of a normally functioning cardiorenal axis is the maintenance of extracellular fluid volume homeostasis. A complex system of volume and pressure sensors, afferent and efferent feedback loops, local and distant vasoactive substances, and neurohormonal systems with built-in redundancies serves to continuously monitor and adapt to changing extracellular fluid volume and blood pressure. When these systems are intact and function normally, they respond rapidly to constantly changing hemodynamics and volume status to ensure adequate tissue perfusion and oxygen delivery. The essential effector mechanisms are the SNA and RAAS. When significant cardiac dysfunction occurs, declining CO and consequent reduction in blood volume in the renal arterial circulation trigger activation of both SNA and RAAS [84]. It has long been recognized that the kidneys of patients with HF release substantial amounts of renin into the circulation [85], which, in turn, leads to increased A II production. By binding to AT1 receptor, A II has broad-reaching effects, including vasoconstriction-mediated increase in systemic vascular resistance, venous tone, and congestion. In addition, A II has potent central nervous system effects including increased thirst, SNA activation and non-osmotic release of vasopressin. In the kidney, A II increases the already high proximal tubular sodium reabsorption and, through preferential constriction of the efferent arteriole, glomerular FF. The latter, as described earlier, increases the oncotic pressure in PC, thus facilitating further return of sodium and water into the circulation [86].
Elevated CVP may have an especially important role in WRF in patient with HFpEF and hypertension. Indeed, in a canine model, renal venous hypertension, independent of changes in systemic arterial blood pressure, led not only to decreased renal blood flow and GFR but also to increased renin release [87, 88]. This provides further evidence that, in HF, renal abnormalities can be caused by neurohormonally induced venous hypertension and congestion in the absence of a decrease in effective circulating blood volume. In addition, the chronic activation of the SNS and RAAS may also contribute to the progression of preexisting CKD. Intrarenal levels of NE, A II, albuminuria, renal function, podocyte injury, and reactive oxygen species production were examined in an animal model of chronic volume overload, created by surgically induced aortic regurgitation in uninephrectomized rats [89]. Chronic volume overload led to predictable changes in the cardiac structure and function with associated increases in both intrarenal SNA and RAAS activity. Importantly, progressive kidney injury could be prevented by renal denervation and A II receptor blockade. Based on these findings, it is plausible that SNA and local A II activation stimulate NADPH oxidase–dependent reactive oxygen species generation in the kidney, which, in turn, causes podocyte injury and albuminuria [89].
Another effect of A II production is stimulation of release from the adrenal gland of aldosterone, which further augments sodium reabsorption in the distal nephron, aggravating pressure and volume overload. Aldosterone has also been implicated in progression of CKD and renal fibrosis [90]. Increased renal aldosterone levels promote oxidative stress. Through paracrine glycoprotein galectin-3 signaling, upregulation of the pro-fibrotic cytokine transforming growth factor-β (TGF-β) leads to increased fibronectin, which promotes renal fibrosis and glomerulosclerosis [91]. In Dahl salt-sensitive HF rats, the combination of ACE and aldosterone inhibition prevented histologic renal damage and lowered both creatinine and proteinuria to control levels. These findings suggest interplay of hypertension-induced and HF-associated renal injury with a related and mutually perpetuating pathophysiology. Inflammation is another non-hemodynamic mechanism contributing to the progression of CKD in the setting of HF [91].
The importance of SNA and RAAS activation in the HF clinical setting is underscored by the unquestionable benefits of RAAS antagonists and beta-blockers which are now the backbone of guidelines-directed medical therapy (GDMT) in HF patients. A detailed description of the studies bringing neurohormonal antagonists to the forefront of HF therapy is beyond the scope of this chapter. However, some aspects of these studies which are especially relevant to the CRS type 2 deserve mention. In the SOLVD study of enalapril in chronic HF, the net deterioration of eGFR from baseline to 14 days was slightly greater in the enalapril group compared to placebo. While early worsening of renal function was associated with increased mortality in the placebo group, it was free from adverse prognostic significance in the enalapril group [92]. Similarly, diabetic patients showed a decreased proteinuria with enalapril treatment [93]. An additional multivariable analysis suggested that despite a higher incidence of early worsening renal function in the enalapril group, there was no risk of longer term deterioration of eGFR compared with placebo [93–97]. Similar findings emerged from studies of aldosterone antagonists and beta-blockers [98]. Nevertheless, the role of neurohormonal antagonists in the prevention of the progression of CRS type 2 remains unclear.
It is important to note that a slight, expected, increase in creatinine, particularly in trials with inhibitors of RAAS, does not necessarily mean a progression of the CRS type 2.
Cardiac resynchronization therapy can improve hypoperfusion in HF as indicated by an increase in GFR by 2.7 ml/min in patients with GFR between 30 and 60 ml/min [99]. The use of LVADs has been shown to improve renal function early after device implantation. The reasons why this improvement appears to be transient have not been elucidated [100].
10.6.3 Cardiorenal Syndrome Type 3
The cardiorenal syndrome type 3, also called the acute renocardiac syndrome, is defined as an episode of AKI which precipitates and contributes to the development of acute cardiac injury and/or dysfunction [65]. There is limited data available describing the role of neurohormonal activation, specifically the SNS and RAAS in the pathophysiology of CRS type 3. However, activation of the SNS is a hallmark of both AKI and acute HF [65]. The enhancement of renal SNS activity and its consequent effect on NE spillover from nerve terminals during AKI [101] may impair myocardial function through several mechanisms, including direct effects of NE, disturbances in myocardial Ca2+ homeostasis, increase in myocardial oxygen demand which increases the risk of subendocardial ischemia, cardiomyocyte apoptosis mediated through β1-adrenergic receptor stimulation, stimulation of α1-adrenergic receptor–mediated cardiomyocyte hypertrophy, and direct RAAS activation [102]. Heightened adrenergic drive can stimulate β1-adrenergic receptors in the juxtaglomerular apparatus of the kidneys contributing to reduced renal blood flow and heightened rennin release and further RAAS activation. Maladaptive RAAS activation in AKI contributes to A II release, vasoconstriction, and further impairment of extracellular fluid homeostasis. In addition, A II contributes to vasoconstriction-mediated increase of systemic vascular resistance. It is also known that A II can directly modify myocardial structure and function, contribute to myocyte hypertrophy, and precipitate apoptosis in cardiomyocyte cultures [103–105]. Furthermore, A II is a potent stimulator of a number of cell signaling pathways including those involved in oxidative stress, inflammation, and the regulation of the extracellular matrix [105]. In a dog model of renal ischemia/reperfusion injury, increased RAAS activity was implicated in the observed reduction of coronary responsiveness to acetylcholine, adenosine, bradykinin, and L-arginine. In addition, renal ischemia/reperfusion injury was associated with increased myocardial oxygen consumption at rest. While a definitive role for the RAAS was conclusively shown, these findings imply that AKI may directly contribute to impaired coronary vasoreactivity and elevated myocardial oxygen consumption both of which can potentially increase the risk of myocardial ischemia and major cardiovascular events [105–110] (Fig. 10.5).
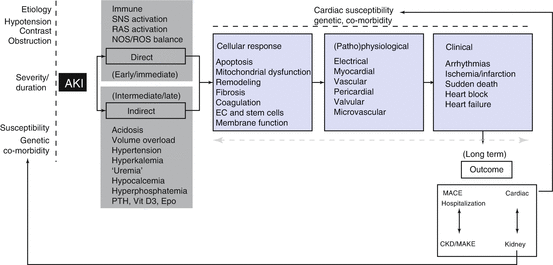
Fig. 10.5
Summary of the demographic contributors, clinical susceptibilities, and pathophysiologic mechanisms for development of CRS type 3. AKI acute kidney disease, SNS sympathetic nervous system, RAS renin angiotensin system, NOS nitric oxygen synthase, PTH parathyroid hormone, Vit D3 vitamin D3, Epo erythropoietin, EC erythropoietic cells, CKD chronic kidney disease (Reproduced from Acute Dialysis Quality Initiative 10, under the terms of the Creative Commons Attribution License. Available at: www.ADQI.org. Accessed 2 July 2015)
10.6.4 Cardiorenal Syndrome Type 4
This type of CRS occurs when CKD (e.g., chronic glomerular disease) contributes to decreased cardiac function, cardiac hypertrophy, and increased risk of adverse cardiovascular events.
The multiple and complex mechanisms which produce mutually detrimental interactions between the kidney and the heart in the CRS type IV are beyond the scope of this chapter and the discussion is limited to the potential roles of neurohormonal activation. However, it is important to note that the risks for cardiovascular complications in patients with eGFRs <30 ml/min/1.73 m2 are up to tenfold higher than those with eGFRs >60 ml/min/1.73 m2 [111]. This startling prevalence of cardiovascular complications exceeds the risk expected from typical risk factors, such as hypertension and hyperlipidemia, and suggests that the loss of renal function may directly contribute to the development of cardiovascular complications [111–115].
The renal response to impaired GFR can lead to activation of multiple compensatory pathways including upregulation of the RAAS and SNA as well as activation of the calcium-parathyroid axis. These physiologic responses can be due to underlying diseases such as hypertension or diabetes or can be a response to the functional decline of either the heart or the kidney. The loss of renal mass leads to the accumulation of total body sodium and water with the subsequent stimulation of A II and aldosterone production. The resulting hypertension coupled with direct effects of A II and aldosterone on cardiac myocytes accelerates left ventricular hypertrophy and cardiac fibrosis. Pathologic adaptations to increasing wall thickness contribute to a loss of capillary density, secondary myocardial fibrosis, and further compensatory hypertrophy [114]. Moreover, the progressive loss of nephron mass inherent to CKD leads to accumulation of salt and water which secondarily contributes to hypertension and pressure and volume overload. These same conditions contribute to LVH through upregulation of RAAS and subsequent release of pro-fibrotic factors such as galectin-3, TGF-β, and endogenous cardiac steroids [116]. In addition, prolonged periods of hemodynamic stress can induce cardiac remodeling which includes the increased expression of interstitial myofibroblasts, a cell type that is not present in normal myocardium and has high fibrogenic potential.
10.6.5 Cardiorenal Syndrome Type 5
This CRS occurs when an overwhelming insult leads to the simultaneous development of acute kidney injury (AKI) and acute cardiac dysfunction. The CRS type 5 encompasses a wide spectrum of disorders that acutely involve the heart and kidney, such as sepsis and drug toxicity where both the heart and the kidney are involved secondarily to the underlying process [65]. The CRS type 5 may develop in the presence or absence of previously impaired organ function. In contrast to the acute CRS type 5, its chronic counterpart, which occurs, for example in liver cirrhosis, has a more insidious onset and the kidney and cardiac dysfunction may develop slowly until overt decompensation occurs.
An essential feature of sepsis is the dissociation between the systemic circulation and the microcirculation in various organs [117]. Especially in the early phases of sepsis, profound microcirculatory changes can develop, despite apparently normal systemic hemodynamics [116–120]. Microcirculatory changes, such as lower blood flow velocities and heterogeneous perfusion patterns, strongly correlate with morbidity and mortality rates [118]. Sepsis can cause both left and right ventricular dilatation and dysfunction, which renders the heart less responsive to fluid resuscitation and catecholamine treatment [119]. Although cardiac dysfunction during sepsis can become severe enough to resemble cardiogenic shock, in the majority of cases, it can be reversed within 7–10 days [120, 121]. Moreover, as long as intravascular volume is maintained, tachycardia and reduced vascular tone may actually contribute to preserve or even increase CO in many patients. Myocardial blood flow or energy metabolism is not as important as previously thought in the development of depressed cardiac function during sepsis [122], which instead appears to be predominantly caused by myocardial depressant factors, including pro-inflammatory cytokines and components of the complement system [122–126].
In experimental models, it has been shown that, regardless of a normal or hyperdynamic systemic circulation, only animals that developed AKI during sepsis have increased renal vascular resistance. These findings are consistent with those of observational clinical studies [127]. Sepsis also affects central neuronal pathways including the SNA, the RAAS, and the hypothalamus-pituitary-adrenal axis, all of which affect cardiac and/or renal function, as repeatedly pointed out in this chapter. Importantly, the severity of sepsis-induced SNA dysfunction is strongly correlated with morbidity and mortality [128, 129]. The hallmark of increased SNA during sepsis is decreased HRV, which has been shown to be associated with the release of inflammatory mediators, such as IL-6, IL-10, and CRP [129, 130]. Data on kidney abnormalities due to sepsis-related autonomic dysfunction are largely preclinical. Interestingly, in a number of animal models, sepsis-induced changes in renal SNA do not appear directly affect renal blood flow [131]. The RAAS activation during sepsis has been deemed to reflect the body’s attempt to restore and maintain an adequate blood pressure. Although counterintuitive, recent experimental and limited clinical data suggest that RAAS blockade might be beneficial, since RAAS activation has also been implicated in endothelial dysfunction, organ failure, and even mortality during severe sepsis [131–134]. Experimental studies have also shown that RAAS activation has detrimental effects on renal function during sepsis [135–138]. During experimental bacteremia, administration of ACE inhibitors improved creatinine clearance and urine output. Furthermore, during experimental endotoxemia, administration of a selective A II type 1 receptor antagonist improved renal blood flow and oxygenation.
Sepsis also causes complex alterations of hypothalamus-pituitary-adrenal signaling, which in some patients results in severe adrenal insufficiency [139]. This in turn triggers increased production of pro-inflammatory cytokines, free radicals, and prostaglandins as well as inhibition of chemotaxis and expression of adhesion molecules. Indeed, short-term administration of moderate-dose glucocorticoids has been shown to reduce the need for vasopressors and length of stay in the intensive care unit [139–141].
Although no definitive data are available on the cardiorenal crosstalk which occurs during sepsis, some specific pathophysiologic mechanisms appear plausible: a reduced cardiac output can reduce renal perfusion, further aggravating sepsis-induced kidney injury; fluid overload due to AKI can lead to overt HF in an already dilated and hypocontractile heart; finally, AKI-induced metabolic acidosis can impair contractility and increases heart rate, worsening myocardial stress [127–139]. Beside the hemodynamic effects of the failing heart on the renal circulation, there are also cardiac changes due to impaired fluid clearance by the kidney. Furthermore, sound experimental evidence shows that AKI itself leads to distant organ function [142]. In a murine model, AKI was associated to decreased cardiac contractility and apoptosis, which was attenuated by treatment with an anti-TNF drug [143]. Cardiac hypertrophy and an increase in cardiac macrophages have also been demonstrated in the setting of sepsis-related AKI [142, 144]. This has particularly profound effects on the brain, which extend to systemic neuroendocrine responses during sepsis [145, 146]. Maintaining hemodynamic stability and maintaining tissue perfusion are key components for preventing CRS type 5 in the hyperacute phase of sepsis. Although fluid resuscitation is essential in early sepsis, continued administration of high fluid volumes can contribute to circulatory congestion and its deleterious consequences, including the development of CRS type 5 [147–150].
The management of cardiac dysfunction in the hyperacute and acute phases of sepsis requires a careful balance between fluid administration to maintain adequate filling pressures and the use of vasoactive drugs to improve cardiac contractility. Although vasopressors can help to restore blood pressure, their indiscriminate use can decrease CO by increasing afterload, especially if hypovolemia is present. Norepinephrine is the preferred vasoconstrictor (α-adrenergic effects with some inotropic effects via its moderate β-adrenergic effects). This drug, which increases systemic blood pressure but decreases renal perfusion in normal conditions, can increase renal perfusion during sepsis [151]. The use of phosphodiesterase inhibitors should be based on careful consideration of their inotropic effects versus their vasodilatatory actions. Although it is a strong vasoconstrictor, vasopressin should be used cautiously as it may have detrimental effects on cardiac output and splanchnic perfusion [152]. Vasopressin paradoxically increases urine output and possibly creatinine clearance in patients with septic shock [151–155]. However, it remains unclear what the target systemic and intrarenal blood pressures should be to optimize renal function [152]. There is no role for dopamine to improve renal hemodynamics and function [156], and there have been limited studies with fenoldopam [157].
The role of the calcium-sensitizer levosimendan in the prevention of the CRS type 5 during sepsis is unknown [158].
Currently, there are no specific drug-based interventions for renal dysfunction in sepsis. General supportive measures include avoidance of nephrotoxic agents, maintenance of an adequate perfusion pressure, and intervention with dialysis. Diuretics have limited roles in the CRS type 5 [155, 158]. Instead, continuous renal replacement therapy should be considered and implemented early, but further studies are needed to validate this concept (Fig. 10.6).
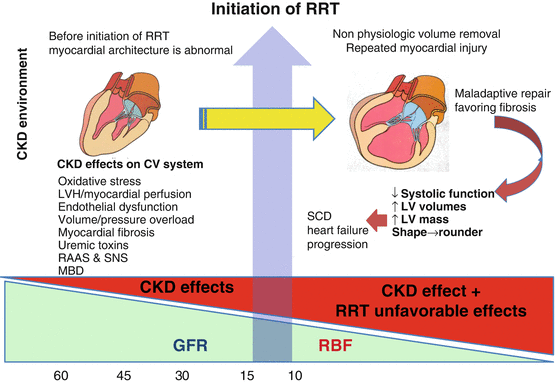
Fig. 10.6
Declining glomerular filtration rate (GFR) is associated with multiple stressors upon the cardiovascular (CV) system including volume/pressure overload, oxidative stress, and activation of the renin angiotensin system (RAAS) and neurohumoral pathways. Prior to the initiation of renal replacement therapy, the heart undergoes maladaptive responses including reduced diastolic compliance, left ventricular hypertrophy (LVH), reduction in myocardial capillary density, and uremia-induced myocardial fibrosis. Following the initiation of renal replacement therapy, “nonphysiologic” fluid removal worsens myocardial ischemia leading to progressive heart failure and sudden cardiac death. CKD chronic kidney disease, SNS sympathetic nervous system, MBD metabolic and bone disorder, RRT renal replacement therapy, RBF renal blood flow, LV left ventricle, SCD sudden cardiac death
10.7 Non-pharmacological Modulation of the Autonomic Cardiorenal Crosstalk in Heart Failure
10.7.1 Renal Denervation
The pathophysiologic mechanisms involved in the autonomic cardiorenal crosstalk raise the question whether renal denervation may produce benefit in HF patients [6]. The discussion which follows will not include studies conducted in resistant hypertension and is rather focused on the available data on the effects of renal denervation in HF [159]. The renal denervation procedure consists of the delivery of low-energy radiofrequency lesions within the renal arteries using electrode catheters positioned just proximal to the origin of the second-order renal artery branch. The technical aspects and potential adverse events of the procedure have been extensively described elsewhere. The ablation of the sympathetic afferent and efferent nerves to the renal arteries should produce a significant reduction in SNA. In HF patients, attenuation of SNA should augment natriuresis, decrease cardiac filling pressures, and potentially improve overall cardiac function [6, 160–162]. Limited data exists on use of renal denervation in HF patients. The principal aim of the Renal Denervation in Patients With Advanced Heart Failure (REACH) pilot study was to examine the safety of renal denervation in a normotensive population with chronic HF [163]. Despite receiving GDMT, the seven study subjects had no hypotension or syncopal episodes while their renal function remained stable over a 6-month period. This very small pilot study also showed a trend toward an improvement in symptoms and exercise capacity [163]. Among 51 NYHA class III and IV HF patients randomized to renal nerves ablation plus optimal medical therapy vs. optimal medical therapy alone, the renal denervation arm had a trend toward reduced HF hospitalizations and improvement in LVEF over a follow-up period of 12 months [164]. These preliminary results are encouraging, but they must be substantiated by larger randomized studies with a longer follow-up. The Renal Denervation in Patients With Advanced Heart Failure (REACH) study (NCT01538992), which was to be a prospective, double-blinded, randomized study on the safety and effectiveness of renal denervation in 100 patients with chronic systolic HF, has been withdrawn prior to enrollment [165]. The Renal Denervation in Patients With Chronic Heart Failure & Renal Impairment Clinical Trial (Symplicity HF) (NCT01392196) is listed on the website clinicaltrials.gov as “active but not recruiting” at the time of this writing [166]. Despite these disappointing events, recent evidence suggesting that renal denervation may reduce left ventricular hypertrophy independent of a drop in blood pressure has raised interest in exploring its role in HF with preserved EF [167]. Pilot studies have also shown that renal denervation may have beneficial effects on glucose metabolism, heart rate, and atrial and ventricular arrhythmias [168–173]. All these comorbidities are characterized by an increased sympathetic tone which is known to adversely affect outcomes in HF patients.
Extreme caution should be used in extrapolating the effects of renal denervation in hypertension studies to those that can occur in an HF population. The long-term impact of renal artery damage is unknown, particularly in HF patients with an already declining renal function. Solid data on the individual variability in renal innervation patterns and in the contribution of the SNA to HF progression are lacking. The pathophysiological implications of renal denervation should be subjected to additional rigorous investigation before this approach can be added to the armamentarium of HF therapies.
10.7.2 Vagal Nerve Stimulation
The vagal nerve originates in the medulla and innervates essentially all the organs in the neck, thorax, and abdomen. The cervical vagal nerve contains both unmyelinated and myelinated nerve fibers. Afferents from the gastrointestinal tract, heart, and lungs outnumber the parasympathetic efferents to the visceral organs. The left vagal nerve gives rise to cardiac efferents that regulate cardiac contractility and the AV node, while efferent fibers in the right vagal nerve act on the sinoatrial node to regulate heart rate [174, 175]. Notably, the mammalian vagal nerve fibers are divided into type A, B, and C. The type of fibers recruited influences the clinical impact of therapies aimed at their modulation. It has been known for more than three decades that there is a strong association between depressed vagal reflexes (as assessed by BRS) and risk of ventricular arrhythmias in the early postinfarction period [176, 177]. Almost two decades ago, the Autonomic Tone and Reflexes After Myocardial Infarction (ATRAMI) study showed that markers of autonomic activity (BRS and HRV) were useful in the risk stratification of patients after a myocardial infarction [178]. Specifically, a depressed BRS in patients with reduced cardiac function identified patients at high risk for HF and arrhythmic mortality [178]. Vagal nerve stimulation (VNS), already used for the treatment of epilepsy and medically refractory depression, is now under investigation also as a treatment for HF [179–183]. The benefit of VNS is due to its central and peripheral antiadrenergic effects and to its anti-apoptotic and anti-inflammatory actions [184]. The role of VNS in HF is supported by the evidence that reduced vagal activity in ADHF is associated with greater hemodynamic abnormalities and increased mortality [182–184]. The improved long-term survival observed in a rodent model of HF induced by myocardial infarction is attributed to the fact that vagal stimulation prevents ischemia-induced loss of connexin 43 thereby improving electrical instability [185]. Additional experimental evidence has shown that VNS improves structural remodeling and EF and reduces a number of inflammatory markers such as TNF-α and interleukin-6 [186].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


