Fig. 2.1
β-adrenergic receptor (AR) signaling in cardiac myocytes. The stimulation of β1-ARs and β2-ARs results in the activation of stimulatory G (Gs) proteins. Gs signaling, in turn, stimulates adenylate cyclase (AC), which converts adenosine triphosphate (ATP) to the second messenger cyclic adenosine monophosphate (cAMP), binding and activating protein kinase A (PKA). PKA can, in turn, phosphorylate and activate several substrates, including l-type calcium channels and phospholamban, ultimately stimulating the increase of free intracellular Ca2+, the regulator of cardiac contractility. β2-ARs also couple to the inhibitory G (Gi) protein and may switch its coupling from Gs to Gi proteins. Differently, β3-AR signaling is associated with nitric oxide release via nitric oxide synthase (NOS) activation
The α1-ARs, instead, couple to the subfamily of Gq heterotrimeric proteins, which activate phospholipase C (PLC)-β. PLC-β catalyzes the formation of the second messengers inositol trisphosphate (IP3) [1, 4, 5]and 2-diacylglycerol (DAG) from the phospholipid phosphatidylinositol-bisphosphate (PIP2). IP3 stimulates the release of Ca2+ from sarcoplasmic reticulum, while DAG activates protein kinase C (PKC), both contributing to increased myocardial contraction and vasoconstriction [1]. Finally, α2-ARs are coupled to the Gi/o family members of the G proteins; thus, its activation inhibits the effector enzyme AC (Fig. 2.2) [22].
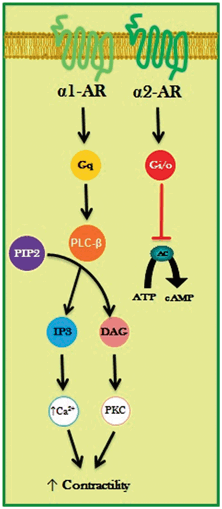
Fig. 2.2
α-adrenergic receptor (AR) signaling in cardiac myocytes. The α1-ARs couple to the subfamily of Gq heterotrimeric proteins, which activate phospholipase C-β (PLC-β) that catalyzes the formation of the second messengers inositol trisphosphate (IP3) and 2-diacylglycerol (DAG) from phosphatidylinositol-bisphosphate (PIP2). IP3 stimulates the release of Ca2+ from sarcoplasmic reticulum, while DAG activates protein kinase C (PKC), both contributing to increased myocardial contractility. The α2-ARs are coupled to the Gi/o family members of the G proteins; thus, its activation inhibits the effector enzyme adenylate cyclase (AC). ATP adenosine triphosphate, cAMP cyclic adenosine monophosphate
Both α2- and β-ARs, like other GPCRs, are subject to complex regulatory mechanisms to protect the receptor from both acute and chronic stimulation, a process termed desensitization. In general, GPCR desensitization involves the following events: (1) receptor phosphorylation and uncoupling from G proteins, (2) internalization of membrane-bound receptors, and (3) downregulation through reduced receptor synthesis or increased degradation of internalized receptors [14].
The desensitization process is orchestrated by three families of proteins: secondmessenger-dependent PKs, GRKs, and arrestins. Initially, GRKs (G-protein-coupled receptors kinases) recognize and phosphorylate agonist-bound receptors, and then they promote the association of cytosolic cofactor proteins called arrestins, which target GPCRs for endocytosis and activate G-protein-independent signaling [23].
The GRKs are a family of cytosolic serine/threonine kinases consisting of seven isoforms with structural and functional similarities. They are classified into three subfamilies: (1) the rhodopsin kinase GRK1 and visual pigment kinase GRK7, (2) the β-AR kinases (or GRK2 and GRK3); and (3) the GRK4 group (GRK4–6). Across mammalian species, GRK2 and GRK5 are the most important members of the GRK family due to their ubiquitous expression; in fact, they are particularly abundant in neuronal tissues and in the heart [24, 25].
GRK2 was first identified as β-AR kinase-1 (β-ARK-1); it is the most expressed GRK subtype in the heart. As a consequence of agonist stimulation, Gβγ subunits interact with GRK2, thus mediating its translocation to the plasma membrane where it initiates receptor desensitization. The importance of GRK2 relies also on the fact that in the last decades, much evidence has suggested a key role for GRK2 in the development of myocardial dysfunction [26, 27] .
Cardiac Adrenergic System in Heart Failure
HF is a chronic clinical syndrome characterized by reduced pumping capacity of the myocardium (systolic HF) , which can develop in response to several cardiac insults [28]. Regardless of the initial cause, the failing heart usually ends up in a vicious cycle of progressive functional decline, with the increased activity of neurohormonal systems aiming to compensate for the reduced blood pressure and cardiac output [1, 29]. In congestive HF, both the activities of the sympathetic nervous system and the renin-angiotensin system (RAS) are increased [30, 31]. In the long term, however, myocardial exposure to high levels of circulating catecholamines and angiotensin increases cardiac workload ultimately leading to maladaptive cardiac remodeling and myocyte death [32, 33].
In HF, ANS hyperactivity is highlighted by increased plasma NE levels, central sympathetic outflow, and NE plasma spillover from activated sympathetic nerve fibers. It has been demonstrated that in patients with HF, cardiac NE spillover is increased by 50-fold, similar to levels found in healthy subjects during maximal exercise [34] . It has also been observed that patients with HF present decreased neuronal density, which can be assessed with 123I-labeled meta-iodobenzylguanidine (MIBG) imaging. MIBG is an analogue of NE, sharing the same uptake, storage, and release processes. As a result, scintigraphic images obtained with MIBG depict the status of catecholamine storage at the level of the myocardial sympathetic presynaptic fibers [35]. Recent MIBG studies have also shown that diabetic patients with HF have lower cardiac sympathetic activity than nondiabetic patients with HF [36].
The ANS hyperactivity observed in HF can also be ascribed to abnormalities in cardiovascular reflexes. In patients with HF, the arterial baroreceptor reflex is largely suppressed, while the sympathoexcitatory reflexes, including the cardiac sympathetic afferent reflex and the arterial chemoreceptor reflex, are augmented [37].
Moreover, other neurohormonal mechanisms, through their interaction with the ANS, can contribute to the progression of cardiac dysfunction in HF. For instance, it has been demonstrated that angiotensin-II can initiate a feedback mechanism leading to the increased sympathetic outflow through the upregulation of the angiotensin-II type 1 receptor and NO inhibition [1] .
From the molecular point of view, typical alterations have been described in failing cardiomyocytes , mainly affecting the adrenergic signaling pathway.
The principal features are the decrease in β1-AR density and mRNA levels, uncoupling of β1-AR from Gs and impaired compartmentalization of cAMP/PKA signaling [38, 39]. As previously mentioned, β-AR desensitization and downregulation are predominantly protective mechanisms, which follow the increase in NE plasma levels with consequent receptor overstimulation. β1-AR abnormalities have been attributed to the recruitment of GRK2 to the agonist-bound receptor [40, 41]. In the past decades, it has been demonstrated that an inverse correlation exists between β-AR density and GRK levels in cardiomyocytes [42], and in transgenic mouse models overexpressing the GRK2 inhibitor β-ARKct, myocardial contractility is increased, and HF development is prevented [43]. In 2005, Iaccarino et al. found an inverse correlation between β-ARs expression and GRK2 levels in patients with HF, with lymphocyte levels mirroring myocardial levels. Interestingly, GRK2 levels also correlated with disease severity, suggesting the importance of GRK2 as a potential biomarker for HF [44]. Recently, the data of a prospective study have been published, indicating that in patients with HF undergoing exercise training, GRK2 levels decrease and are also able to predict survival [45].
Further data on the importance of GRK2 in HF came from preclinical studies using gene therapy approaches. A study was performed in which long-term cardiac gene therapy with an adeno-associated vector encoding for -ARKct resulted in sustained improvement of cardiac function , reversal of remodeling, and normalization of the neurohormonal signaling axis in a rodent model of HF [46].
The role of β2-ARs in HF has not been clearly defined yet. In the failing heart, levels of β2-ARs do not change significantly, and studies in transgenic animals have demonstrated that while only fivefold overexpression of β1-AR results in cardiomyopathy, even a 100-fold increase of β2-ARs in the mouse heart does not have any detrimental consequences but, instead, significantly increases cardiac contractile force [20] . This probably needs to be ascribed to the fact that β2-ARs also signal through Gi proteins, which can activate a protective antiapoptotic pathway. Recently, the role of β2-AR has also been studied in a murine postischemic model of HF. Adenoviral-mediated β2-AR overexpression resulted in improved angiogenesis and enhanced coronary reserve and myocardial blood flow in HF mice. This was associated with the activation of the proangiogenic pathway mediated by the vascular endothelial growth factor (VEGF)/protein kinase B (PKB)/endothelial NOS (eNOS) [47, 48].
The role of β3-ARs in HF has not been elucidated. Some evidence suggest that in HF there is an excess of β3-AR signaling, which exerts a negative inotropic effect by increasing NO production and inhibiting calcium transients [49].
α1-ARs, which are involved in cardiomyocyte growth and pathological hypertrophy, may also play a compensatory role in HF in order to preserve cardiac inotropy. In particular, the α1A-subtype has shown to produce prosurvival effects and to protect from maladaptive remodeling both in models of pressure overload and acute myocardial infarction [50].
As for the role of α2-AR in HF, it has been extensively investigated in studies evaluating the contribution of the adrenal gland function in HF, which will be discussed below .
Adrenergic Signaling in Adrenal Gland During Heart Failure
The ANS hyperactivity has been known as a peculiarity during HF [29, 51]. Sympathetic overdrive is strictly implied in the establishment and development of cardiac dysfunction, determines higher risk of arrhythmias, and contributes to worsen the prognosis in patients with HF (increase in plasmatic concentration of catecholamines leads to significant higher mortality) [52].
Indeed, HF is characterized by elevated sympathetic tone, which involves increased levels of circulating and synaptic catecholamines . Cardiac sympathetic nerve activity is significantly augmented in animal models of HF as well as muscle sympathetic nerve activity in patients with HF [53].
Simultaneously, in the adrenal gland, there is an increase in catecholamine output and secretion attested by higher tyrosine hydroxylase levels that is a key enzyme in the production of both NE and Epi.
Chromaffin cells of adrenal medulla are the major source of circulating catecholamines and secrete into the blood 80 % Epi and 20 % NE [54, 55]. The adrenal gland should be considered a specialized sympathetic ganglion with the distinguishing characteristic to excrete its neurohormones directly into the bloodstream [56]. Catecholamines are secreted from chromaffin cells after acetylcholine stimulation of the nicotinic cholinergic receptors; this secretion is regulated by many receptors, among which we mention β2-ARs and α2-ARs that have, respectively, a facilitatory and an inhibitory role [29].
Recently, some studies have shown the crucial inhibitory role of presynaptic α2-AR in peripheral nerve terminals and in adrenal medulla during HF. In particular, α2A- or α2C-AR knockout (KO) mice that underwent HF after transverse aortic constriction manifested an increase in circulating catecholamines and a worst cardiac function compared with control mice [57], while, more interestingly, double α2A/α2C-AR KO mice developed cardiomyopathy at 4 months of age, without any treatment [58]. Moreover, this finding was corroborated by comparable results in human polymorphisms of α2C-AR: α2CDel322–325 in healthy people determines an increase in ANS activity and in plasmatic catecholamine levels during supine rest and an improved pharmacologically induced NE and Epi secretion; besides, the same polymorphisms associated with high HF risk influence the therapeutic effects of the -blocker bucindolol in patients with HF [59–61].
Few years ago, it has been shown that adrenal GRK2 is a physiological regulator of catecholamine production/secretion [62], and its upregulation is fundamental for α2-AR desensitization/downregulation in animal models of HF [55, 63, 64]. In adrenal gland , overexpressed GRK2 phosphorylates α2-ARs determining their dysfunction and leading to their incapability to inhibit catecholamine production during HF. A gene therapy that inhibits adrenal GRK2 levels (through the peptide β-ARKct) is able to restore α2-AR membrane levels/function and consequently to decrease plasmatic catecholamine levels. Therefore, this sympatholytic therapy contrasts the detrimental cardiac effects of catecholamines and allows re-sensitizing cardiac β-ARs, to decrease left ventricle dilatation and to importantly improve systolic function. Moreover, adrenal GRK2 downregulation has an important role on beneficial sympatholytic effects of β-blockers and exercise training during HF [65, 66].
Hence, since adrenal GRK2 is crucial for α2-AR dysregulation and subsequent increase of circulating catecholamine secretion during HF, its inhibition (through gene therapy, small molecules, or peptides) may represent an innovative sympatholytic therapeutic strategy for HF [67].
Conclusions
Adrenergic system is a key regulator of cardiac activity both in physiologic and in pathological conditions. Indeed, the consequences of sympathetic hyperactivity (and mainly of β-ARs) during HF have been demonstrated to play a fundamental role in the progression of this disease, thus overturning the therapeutic strategies used in the treatment of this and other cardiovascular diseases. Therefore, efforts have been made to further understand adrenergic signaling influencing heart activity, focusing not only on the heart and β-ARs but also on different “players” in the pathogenesis of HF, thus looking at HF as a “systemic” disease involving a dysregulation of various tissues and systems. In this vein, new potential pivotal regulators of sympathetic systems, such as adrenal presynaptic α2-AR or cardiac and adrenal GRK2, have been identified in the last years; such new targets might represent additional, useful tools to increase the therapeutic strategy in the treatment of cardiovascular diseases.
References
1.
2.
Barrese V, Taglialatela M. New advances in beta-blocker therapy in heart failure. Front Physiol. 2013;4:323.CrossRefPubMedCentralPubMed
3.
Lymperopoulos A. Physiology and pharmacology of the cardiovascular adrenergic system. Front in Physiol. 2013;4:240.CrossRef
4.
Granneman JG. The putative beta4-adrenergic receptor is a novel state of the beta1-adrenergic receptor. Am J Physiol Endocrinol Metab. 2001;280:E199–202.PubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


