Fig. 4.1
How the subclassification of adrenoceptors has developed since 1948
Further studies were hampered by the lack of an antagonist at β-adrenoceptors, until Powell and Slater [111] reported dichloroisoprenaline as the first useful β-blocker. Furchgott [51], using α- and β-adrenoceptor antagonists, confirmed and expanded the results of Ahlquist on intestinal smooth muscle, demonstrating that both α- and β-adrenoceptors were involved in the inhibition .
The Ahlquist classification was refined by the identification of two subtypes of β-adrenoceptor, based on the affinities of adrenaline and noradrenaline , where β1-adrenoceptors were mainly cardiac and involved in excitation and stimulated by both adrenaline and noradrenaline , and β2-adrenoceptors were mainly involved in smooth muscle relaxation and the targets of adrenaline [77] (see Fig. 4.1). A third β-adrenoceptor was later identified [42] and shown to be involved in β-adrenoceptor-mediated relaxations resistant to propranolol and termed the β3-adrenoceptor [18] (see Fig. 4.1). A fourth β-adrenoceptor termed β4 and involved in vascular relaxations is now known to be a low-affinity state of the β-adrenoceptor[89] .
The next major development in adrenoceptor classification did not occur until 1974, but the detail does not directly concern vascular adrenoceptors. Basically, agonists and antagonists at α-adrenoceptors were found to reduce or increase, respectively, the release of noradrenaline evoked by nerve stimulation from adrenergic nerves, and evidence accumulated to show that these actions were mediated by α-adrenoceptors on the nerve terminals, so-called presynaptic or prejunctional receptors [78, 127].
Once the concept of prejunctional and postjunctional α-adrenoceptors had been accepted, differences between pre- and postjunctional α-adrenoceptors pharmacologically, in terms of the relative potencies of a series of agonists and antagonists, led to the further subclassification of α-adrenoceptors into α1-postjunctional and α2-prejunctional. Later, it became clear that α2-adrenoceptors may also be present postjunctionally, so that this classification was expanded into a purely pharmacological subclassification, independent of location [10] (see Fig. 4.1) .
Further refinements in the classification of adrenoceptors have come from the development of new methodologies for the study of receptors. The first of these was the radioligand-binding assay which, beginning in the mid-1980s, began to demonstrate that there were subtypes of α1-adrenoceptors [61, 97], α2-adrenoceptors [17] and β-adrenoceptors [42]. However, the relationship between affinity at ligand-binding sites and potency at functional receptors was not always easy to determine [32]. The study of adrenoceptors was revolutionised by the techniques of molecular biology, which definitively defines receptors as gene products. Nine genes for adrenoceptors have now been identified and sequenced (α1A, α1B, α1D; α2A, α2B, α2C; β, β, β) and species orthologues have been identified allowing simplification of classification (human α2A and rat α2D ligand-binding sites are now simply classed as α2A) [18]. Studies of subtypes of receptor have been made easy by transfection of genes into suitable cell lines to produce pure populations of recombinant receptors.
The object of this chapter is to look at the physiological function of subtypes of adrenoceptors in the vascular system, looking at each subtype individually. Figure 4.1 shows the historical developments in our knowledge of the subtypes of adrenoceptor, and the nine established subtypes of adrenoceptor [see 34] .
Smooth Muscle α1-Adrenoceptor Subtypes
α1-Adrenoceptors were initially subdivided into α1A and α1B subtypes in ligand-binding studies, based on the affinities of a series of ligands, especially WB 4101 and prazosin [97], and based on the ability of the alkylating agent chloroethylclonidine to inactivate the α1B but not the α1A subtype [61].
Molecular cloning techniques revealed initially four subtypes of α1-adrenoceptor [18]. The α1B-adrenoceptor subtype was the first to be cloned, from the hamster [26], and this clone was so named because it expressed a protein with the radioligand binding properties of the α1B-adrenoceptor. Other clones were rat α1A [85], bovine α1C [118] and rat α1D [108]. However, whereas the α1C clone represented the α1A-ligand-binding site, the α1A and α1D clones appeared to represent the same subtype: a novel subtype of α1-adrenoceptor that was termed to avoid confusion, α1D. These clones have now been renamed to match the functional receptors: α1A (formerly α1C), α1B (formerly α1B) and α1D (formerly α1A/α1D).
Vascular Responses Mediated by α1A-Adrenoceptors
Contractions are reported to be mediated at least partly by α1A-adrenoceptors in a number of tissues including rat mesenteric artery (see Fig. 4.2), rat renal artery (also α1D) [141], rat-tail artery [76, 140] and rabbit ear artery [45]. See also the following section on α1D-adrenoceptors.
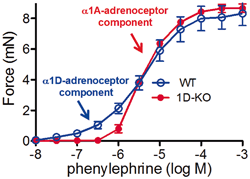
Fig. 4.2
Concentration–response curves for contractions to the α1-adrenoceptor agonist phenylephrine in small mesenteric artery from wild-type (open circles) and α1D-adrenoceptor knockout (KO) mice (filled circles). The α1D-adrenoceptor-mediated response is the component found in wild-type but not in α1D-adrenoceptor KO mice. Note that low concentrations of phenylephrine (and indeed noradrenaline) selectively activate α1D-adrenoceptors. WT wild type. (Taken from Bexis and Docherty, unpublished)
α1L-Adrenoceptors
α1-Adrenoceptors in blood vessels were subdivided based on their affinities for prazosin, WB 4101 and HV 723 into α1H and α1L [100]. α1H-Adrenoceptors had high affinity for prazosin, and appeared to match the α1A, α1B, α1D classification [101], whereas α1L had low affinity for prazosin and did not seem to match the current molecular-cloning-based classifications. Under this classification, and often based on the low potency of prazosin, rabbit aorta, mesenteric and carotid arteries [100], guinea pig aorta, [101], rabbit cutaneous resistance arteries [123] and rat small mesenteric artery [137] were reported to contain α1L-adrenoceptors.
Knockout (KO) of the α1A-adrenoceptor abolishes α1L-adrenoceptor pharmacology. The α1A-adrenoceptor, expressed in the Chinese hamster ovary CHO-K1 cell line, displayed binding properties of the α1A-adrenoceptor, but functionally, in terms of inositol phosphate accumulation, the receptor displayed properties of the α1L-adrenoceptor [49]. Hence, the same receptor showed characteristics of both α1A– and α1L-adrenoceptors: in ligand-binding studies α1A, but in functional studies α1L. However, receptors reported to be α1L-adrenoceptors can be demonstrated under slightly altered conditions to be α1A-adrenoceptors. Hence, in the view of this author, it is likely that the α1L-adrenoceptor is simply the native α1A-adrenoceptor, at which prazosin shows low potency functionally [37] .
Vascular Responses Mediated by α1B-Adrenoceptors
The α1B-adrenoceptor has been somewhat a mystery receptor in vascular studies. Contractions are reported to be mediated at least partly by α1Β-adrenoceptors in a number of tissues: rabbit corpus cavernosum [104] and rabbit cutaneous resistance arteries [123]. In studies of blood vessels from adrenoceptor KO mice, it was found that the major subtypes involved in contractions are α1A and α1D, but that the α1Β-adrenoceptor plays a minor but clearly defined role in contractions in aorta, carotid, mesenteric and tail arteries [28]. For instance, contractions in rat-tail artery develop more slowly in α1Β-adrenoceptor KO mice [28], so that subtle differences can be revealed following receptor KO. In the mouse mesenteric vasculature, α1B-adrenoceptor KO resulted in diminished nerve but not agonist-induced vasoconstriction at 10 Hz but not 2 or 4 Hz [136].
Vascular Responses Mediated by α1D-Adrenoceptors
BMY 7378 is a selective antagonist at α1D-adrenoceptors [58] and has been useful in identifying responses mediated by α1D-adrenoceptors. Contractions are reported to be mediated at least partly by α1D-adrenoceptors in a number of tissues including: rat aorta [1, 69, 110], rat iliac artery [110], rat mesenteric artery and pulmonary artery [69], rat renal artery (also α1A) [141], rat carotid artery, mesenteric artery, aorta [140] and rabbit aorta (also possibly α1A) [45]. In contrast, some studies of rat mesenteric artery did not report α1D-adrenoceptor involvement in contractions [110]. The α1D-adrenoceptor is reported to be constitutively active in the rat aorta, meaning that a response can occur in the absence of an agonist, and antagonists such as prazosin behave not simply as competitive antagonists but also as inverse agonists to inhibit that constitutive activity [6, 57].
Vascular Responses Mediated by Multiple Subtypes of the α1-Adrenoceptor
Clearly, contractions in a number of tissues are mediated by more than one subtype of the α1-adrenoceptor, and currently available subtype selective antagonists are often not selective enough to tease out clearly which receptors are present. In the perfused rat kidney, both α1A– and α1D-adrenoceptors mediate vasoconstriction [5], and in the external carotid circulation, the use of selective antagonists suggests that responses involve α1A, α1D, α2A and possibly α2C [143a]. Unlike the rat aorta, where the predominant receptor is α1D, contractions of the mouse aorta clearly involve α1A– in addition to α1D-adrenoceptors [146]. Receptor KO studies in mice can help. In α1D-adrenoceptor KO mice, there was a reduced vasoconstrictor response to both NA and nerve stimulation in femoral arteries [148], demonstrating the role of this receptor in neurotransmission (see later). In mouse mesenteric arteries, there is evidence from antagonist studies for the involvement of both α1A– and α1D-adrenoceptors in contractions [146], although the predominant α1-adrenoceptor is the α1A-adrenoceptor.
The role of the α1A– and α1D-adrenoceptor in contractile responses in a number of blood vessels may best be illustrated by Fig. 4.2. Figure 4.2 shows concentration–response curves for contractions produced by the α1-adrenoceptor agonist phenylephrine in small mesenteric artery from wild-type (WT) and α1D-adrenoceptor KO mice. KO of the α1D-adrenoceptor changes the response from a relatively shallow concentration–response curve to a steep curve. The α1D-adrenoceptor-mediated response is the component found in WT but not in α1D-adrenoceptor KO mice. Note that low concentrations of phenylephrine (and indeed noradrenaline) selectively activate α1D-adrenoceptors (Fig. 4.2). The high-affinity receptor is an α1D-adrenoceptor. This is also true for noradrenaline: Noradrenaline has high potency at the α1D-adrenoceptor and lower potency at the α1A-adrenoceptor, so that knockout of the α1D-adrenoceptor removes the high-potency component of the response to noradrenaline . The high affinity of this receptor for noradrenaline makes it the most likely candidate for the receptor in the neuroeffector region involved in the control of neurotransmission, and indeed blood pressure is reduced by deletion of this receptor (see below).
The suggestion that noradrenaline has higher potency at α1D-adrenoceptors is confirmed in many pharmacological studies. Indeed, noradrenaline has been reported to have high potency (aorta: pEC50 of 8.15) [24] in tissues with a high level of α1D-adrenoceptor messenger RNA (mRNA; aorta: 70–80 %; [89a]) and low potency (pEC50 of 6.32) [126] in the small mesenteric artery which had a high level of α1A-adrenoceptor mRNA (75 %; [89a]).
In mouse mesenteric arteries, where, as discussed above, the predominant receptor is α1A-adrenoceptor (see Fig. 4.2), prazosin potency (pK B, −log M) was 8.8 in WT mice, but 9.6 in α1B-/α1D-adrenoceptor KO mice [91]. However, prazosin pK B was 10.3 in α1A-/α1B-adrenoceptor KO mice [92]. In another study, prazosin pA 2 was 9.92 in WT and 9.83 in α1B KO, but 9.30 in α1D KO and 9.43 in α1B/α1D KO [67]. These studies seem to agree that prazosin may be more potent in the mouse than in the rat, but there is no conclusive evidence that its potency is decreased by α1DKO.
Physiological Blood Pressure Responses Mediated by α1-Adrenoceptors
Piascik et al. (1995) [110] reported that the α1A-adrenoceptor subtype plays a role in the tonic maintenance of blood pressure in the conscious rat, whereas the α1B-adrenoceptor subtype participates in the response to exogenous agonists. In receptor KO mice, α1A-adrenoceptor KO significantly reduced resting blood pressure in only two from four studies, and indeed a combined α1A-/α1B-adrenoceptor KO failed to affect basal blood pressure in two studies (see Table 4.1), α1B-adrenoceptor KO did not affect resting blood pressure, but α1D-adrenoceptor KOs significantly reduced resting blood pressure in three from three studies (Table 4.1). In KO mice lacking the α1B-adrenoceptor subtype, there was no effect on basal blood pressure, but pressor responses to phenylephrine were significantly blunted [21]. However, overexpression of α1B-adrenoceptors results in hypotension and cardiac hypertrophy, suggesting that effects seen in α1B-adrenoceptor KO mice may not relate to direct blood pressure actions of α1B-adrenoceptors (otherwise, overexpression would result in increased blood pressure) [151]. In the pithed rat, a component of the vasopressor nerve response, previously identified as α2-adrenoceptor mediated based on the effects of the α2-adrenoceptor antagonist yohimbine, has been reclassified as α1D-adrenoceptor mediated [35]. Yohimbine was found to have unexpected α1D-adrenoceptor potency. Hence, in the pithed rat, pressor nerve responses may involve both α1A– and α1D-adrenoceptors [35].
Table 4.1
Baseline blood pressure in wild-type and α1– and α2-adrenoceptor KO mice
(a) α 1 -Adrenoceptor KO mice | |||||
WT | α1A KO | α1B KO | α1D KO | Notes and double KO | Reference |
114 | 104* | Male | [115] | ||
111 | 102 | 99 | 93* | Female | [115] |
99 | 99* | 1B/1D KO: 92* | [67] | ||
109 | [131] | ||||
111 | Male 1A/1B KO: 112 | [105] | |||
111 | Female 1A/1B KO: 111 | [105] | |||
120 | 111 | [136] | |||
105 | 102 | 104 | 95* | 1A/1B KO: 104 | [68] |
(b) α 2 -Adrenoceptor KO mice | |||||
WT | α2A KO | α2B KO | α2C KO | Reference | |
120 (est) | 120 (est) | [84] | |||
119 | 118 | [21] | |||
128 | 131 | [4] | |||
111 | 118* | [102] | |||
91 | 97 | 2A/2C KO: 114* | [56] | ||
146 | 155* | [150] | |||
These results may suggest that α1A– and α1D-adrenoceptors and perhaps α1B-adrenoceptors are involved in blood pressure control . However, the clearest result in terms of basal blood pressure is with the α1D-adrenoceptor KO mouse: Blood pressure has been consistently reported to be significantly lowered in α1D KO mice (see Table 4.1).
Smooth Muscle α2-Adrenoceptor Subtypes
α2-Adrenoceptors have been subdivided into three subtypes, α2A-, α2B– and α2C-adrenoceptors, based on ligand-binding and molecular cloning studies [18], and the α2D-adrenoceptor initially found in the rat is a species orthologue of the human α2A-adrenoceptor, so that this subtype nomenclature has been dropped. The important features of α2-adrenoceptor subtypes are shown in Table 4.2.
Table 4.2
Summary of vascular α2-adrenoceptor subtype characteristics
Receptor subtype | 2A | 2B | 2C |
|---|---|---|---|
Functional responses (vascular) | Vascular contraction: minor role? | Vascular contraction | Vascular contraction: human saphenous v |
Functional responses (blood pressure) | Central control of blood pressure: central hypotension, peripheral pressor | Control of blood pressure: peripheral pressor | Minor?? |
Affinity of NA? | |||
Selective agonists | |||
Non-selective antagonist | Yohimbine | Yohimbine | Yohimbine |
Selective antagonists | BRL44408? | ? | |
G protein | Gi | Gi | Gi |
Second messenger | Inhibit AC | Inhibit AC | Inhibit AC |
Postjunctional α2-Adrenoceptors In Vivo
Following the identification of prejunctional α2-adrenoceptors, evidence was presented, employing α2-adrenoceptors antagonists and especially yohimbine, that α2-adrenoceptors may be present on vascular smooth muscle and to mediate vasoconstrictor responses, in terms of actions both of exogenous agonists and of endogenous neurotransmitters [39, 40, 41]. More recently, the subtypes of α2-adrenoceptor involved in these pressor responses to exogenous agonists were investigated and it was found that the predominant receptor was α2A-adrenoceptor [54]. However, it has since been established that the component of the vasopressor nerve responses in the pithed rat found to be blocked by yohimbine are actually α1D-adrenoceptor mediated [35]. Fortunately, there is indeed an α2A-adrenoceptor-mediated component to pressor responses to exogenous agonists in the pithed rat [36].
Physiological Blood Pressure Responses Mediated by α2-Adrenoceptors
The effects of α2-adrenoceptor receptor KOs on resting blood pressure have been examined: The only change in resting blood pressure produced by α2-adrenoceptor KO is an increase in blood pressure in α2A-adrenoceptor KO mice (see Table 4.1). The major effect of KO of the α2A-adrenoceptors is abolition of a central hypotensive action [102]. As has been elegantly shown using KO mice, α2A-adrenoceptors are involved in the central hypotensive actions of α2-adrenoceptor agonists and hence the biphasic transient pressor and prolonged depressor response to the α2-adrenoceptor agonist UK14,304 in WT mice was altered to a transient (but more prolonged) pressor response in α2A-adrenoceptor KO mice [87, 88]. Hence, the crucial α2-adrenoceptor in terms of blood pressure control is the α2A-adrenoceptor, involved presumably in tonic central control of blood pressure (although a component due to peripheral effect at prejunctional α2A-adrenoceptors on sympathetic nerves innervating blood vessels cannot be ruled out).
There were no obvious haemodynamic effects produced by disruption of the α2C-adrenoceptor [84]. However, the biggest mystery surrounds the α2B-adrenoceptor. Knockout of the α2B-adrenoceptor abolished the early transient pressor response to the α2-adrenoceptor agonist dexmedetomidine and prolonged the depressor response suggesting that the early pressor response is α2B-adrenoceptor mediated [84]. These effects were admittedly obtained with a single dose of α2-adrenoceptor agonist. These blood pressure effects are systemic cardiovascular responses and do not allow for subtle effects on different vascular beds, nor do they prove categorically that the α2A-adrenoceptor subtype has purely central actions or that the α2B-adrenoceptor subtype is the only mediator of pressor responses. It is possible that α2A-adrenoceptors mediate both central hypotension and peripheral hypertension, but the central actions dominate in conscious animals.
The receptor knockout studies would suggest that the α2B-adrenoceptor and not the α2A-adrenoceptor is involved in peripheral pressor responses or at least that the α2B-adrenoceptor is the predominant receptor involved under the experimental conditions and with the chosen agonist. How do these findings on blood pressure in KO mice relate to the above studies in the pithed rat in which pressor responses were mediated by α2A-adrenoceptors (see above)? The pithed rat results do not rule out the involvement of α2B-adrenoceptors in pressor responses in pithed rats, but suggest that α2A-adrenoceptors are dominant. The identification of α2A-adrenoceptors relies on BRL 44408, but no reliable selective α2B-adrenoceptor antagonist is available. The lack of really good selective α2B– or indeed α2C-adrenoceptor antagonists hinders pharmacological studies. We need more selective antagonists to answer these questions.
Hence, in general, it seems that the major contributors to exogenous-agonist-induced pressor responses in vitro and to exogenous-agonist-induced contractions in vitro are α1A-, α1D– and α2A (or α2B)-adrenoceptors. However, effects of exogenous agonists, while of importance in terms of pharmacological actions of drugs, may tell us little about what receptors are important in the physiological control of the vasculature. Hence, KO studies can be of use in clarification of this point.
α2-Adrenoceptors in Isolated Tissues
In terms of postjunctional α2-adrenoceptors in isolated tissues, there is relatively little evidence available as to subtypes mediating vascular contractions , given the relatively few isolated tissue preparations in which these receptors can be demonstrated. The human saphenous vein is a preparation in which it appears that the dominant adrenoceptors mediating contractions are α2-adrenoceptors. In studies from this laboratory of side branches of the human saphenous vein, there appears to be a homogeneous population of α2-adrenoceptors with no evidence for α1-adrenoceptors : Yohimbine was more potent than prazosin; both yohimbine and prazosin produce parallel shifts in the noradrenaline concentration–response curve [see 122]; other α1-adrenoceptor antagonists such as HV 723 [100] had relatively low potency; the α1-adrenoceptor selective agonist phenylephrine had low potency; and the α2-adrenoceptor selective agonist oxymetazoline had high potency [55]. Other studies of the human saphenous vein have reported that contractions are mediated predominantly by α2-adrenoceptors [99, 128], or by α1– and α2-adrenoceptors [43, 114]. However, it is important to note that α2-adrenoceptors identified employing yohimbine at a time before the functional identification of the α1D-adrenoceptor must be treated with caution until selective α1D-adrenoceptor antagonists are also examined: Yohimbine has actions at α1D-adrenoceptors in concentrations only five- to tenfold less than those at which it has actions at α2-adrenoceptors. Hence, some of these results have been reconsidered in terms of possible α1D-adrenoceptor involvement: In the human saphenous vein, responses to exogenous noradrenaline have been shown to be α2C– [55] and not α1D-adrenoceptor mediated [24].
In the porcine palmer lateral vein and common digital artery, the low potency of prazosin suggests the presence of α2A-adrenoceptors [12], and α2A-adrenoceptors have also been reported in dog saphenous vein [86]. In the pig ciliary artery, the high potency of BRL44408 suggests the involvement of α2A-adrenoceptors in contractions [143]. In a number of tissues, α2-adrenoceptors contribute to a predominantly α1-adrenoceptor-mediated response. In rat cremaster arterioles and venules, a component of the contractile response, based on the potency of BRL 44408, seems to be mediated by α2A-adrenoceptors [83].
Initially, it was suggested that vascular α2-adrenoceptors may be predominantly extrasynaptic and mediate responses to circulating catecholamines [79], but it was later found that contractions to nerve stimulation in the human saphenous vein are α2-adrenoceptor mediated, based on high potency of yohimbine and low potency of prazosin [38]. The low potency of prazosin rules out an important role for α1D-adrenoceptors in this response.
Smooth Muscle β-Adrenoceptor Subtypes
Although, in the initial classification by Lands et al. [77], it was the β2-adrenoceptor that mediated vascular relaxations , it was not long before evidence that both β1- and β2-adrenoceptors were involved in vascular relaxations was obtained [106]. Soon after, β2- [30] and then β1-adrenoceptors [50] were cloned. A third β-adrenoceptor was identified [42] and shown to be involved in β-adrenoceptor-mediated relaxations relatively resistant to propranolol, and termed the β3-adrenoceptor [18]. The important features of β-adrenoceptor subtypes are shown in Table 4.3. Information on selective agonists and antagonists is listed in Table 4.3 [7].
Table 4.3
Summary of vascular β-adrenoceptor subtype characteristics
Receptor subtype | ß1 | ß2 | ß3 |
|---|---|---|---|
Functional responses (vascular) | Relaxation. Neurotransmission target? | Widespread relaxation. Target of adrenaline | Relaxation |
Affinity of NA? | High | Low | Low |
Selective agonists | Xamoterol | Formoterol | ? BRL37344 |
Non-selective antagonist | Propranolol | Propranolol | propranolol (low potency) |
Selective antagonists | atenolol, CGP 20712A | ICI 118551 | SR59230A |
G protein | Gs | Gs | Gs |
Second messenger | Stimulate AC | Stimulate AC | Stimulate AC |
β-adrenoceptor-mediated dilatation can involve a number of possible actions:
a.
Direct vascular smooth muscle relaxation due to activation of adenylate cyclase and increase in cyclic adenosine monophosphate (cAMP) production. cAMP activates protein kinase A (PKA) which causes protein phosphorylation linked to vascular relaxation.
b.
Endothelium-dependent relaxation involving nitric oxide production and stimulation of guanylate cyclase and increase in cyclic guanosine monophosphate (cGMP).
c.
Hyperpolarization of endothelium or smooth muscle cells.
In a comparison of rat mesenteric artery and aorta, it was found that all three β-adrenoceptors were present in both vessels, but in mesenteric artery vasodilatation was mediated by β1-adrenoceptors on vascular smooth muscle via cAMP production (but with additional β3-adrenoceptors on endothelium and smooth muscle but perhaps not involved in relaxation), and in aorta was mediated by β2-adrenoceptors on both the endothelium and smooth muscle and by β3-adrenoceptors on the endothelium [48a].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


