Fig. 3.1
Schematic representation of β2-AR signaling to eNOS and the role of the Gi–Src–Akt axis. See text for details and molecular acronym definitions. cAMP cyclic adenosine monophosphate, PKA protein kinase A, β 2 -AR β2adrenergic receptors, MAPK mitogen-activated protein kinase, eNOS endothelial NO synthase
There is also evidence for the functional presence of vasorelaxant α1-AR in the brachial and pulmonary arteries isolated from rabbit and rat, respectively [13, 68]. According to these reports, the pharmacological stimulation of α1-AR located on ECs, is able to generate NO, whereas the stimulation of α2-AR releases a relaxing prostanoid [13, 68]. Filippi et al. demonstrated that nanomolar concentrations of phenylephrine, which are devoid of any contractile effect, induced a slight endothelium-dependent vasorelaxation in the rat mesenteric vascular bed through the stimulation of α1D-AR, located on ECs, which act through phospholipase C stimulation, followed by IP3 generation and NO synthase activation. Conversely, the increase in perfusion pressure induced by micromolar concentrations of phenylephrine is attributable to the stimulation of α1A-AR [69]. α2-AR agonists cause endothelium-dependent relaxation that is reduced or abolished by inhibitors of L-arginine/NO. The activation of α2-AR on ECs stimulates the release of NO, an action that would tend to attenuate vasoconstriction produced by the activation of postsynaptic vascular α1-ARs [19, 20, 70]. The α2-AR subtype that causes endothelium-dependent relaxation is the α2A/D subtype, despite the prominent presence of α2C-ARs (77 α2C vs. 23 % α2A/D) [22]. It appears that this ratio may not be constant, since it varies within the vascular bed. Indeed, Bockman et al. demonstrated that, in the rat mesenteric artery, the α2-AR is coupled to endothelium-dependent NO-mediated relaxation and is of the α2A/D subtype (appearing in its α2D version) [21]. It has been demonstrated that endothelium-dependent relaxation to α2-adrenergic agonists is prevented by pertussis toxin [24, 71–75], suggesting the involvement of Gi proteins in the signal transduction from the receptor to the activation of NO synthase [14, 76]. Indeed, α2-adrenergic agonists cause the activation of Gi proteins in ECs and stimulate NO synthase activity [77, 78]. Surprisingly, cAMP is not involved in the signal transduction pathway of α2A/D-AR-mediated NO formation [21]. Indeed, the use of forskolin to oppose α2-AR-mediated inhibition of cAMP formation in endothelium did not affect the relaxant response to α2-AR agonists, suggesting that cAMP is not involved in the coupling of α2-AR to NO. There is a physiological modulation of endothelium-dependent relaxation to α2-adrenergic agonists. Such relaxation is upregulated by a chronic increase in blood flow [79] or exercise training [80].
Adrenergic System and Angiogenesis
Vascular growth is a complex process involving both angiogenesis (creation of new capillaries) and arteriogenesis (enlargement and remodeling of preexisting collaterals) [81]. More specifically, the term angiogenesis refers to the sprouting, enlargement, or intussusceptions of new endothelialized channels and is tightly associated to ECs proliferation and migration in response to angiogenic stimuli, in particular hypoxia. Arteriogenesis is, instead, a result of growth and positive remodeling of preexisting vessels, forming larger conduits and collateral bridges between arterial networks via recruitment of smooth muscle cells. In particular, angiogenesis has long been known to be a highly ordered multistep molecular process under tight regulation by ECs [82] and closely associated with EC proliferation and migration and to the capability of these cells to modulate the levels of VEGF, the most important cytokine system involved in the formation of new vessels [83]. However, a series of biological, chemical, and hormonal effectors can interfere with this process, including the adrenergic system through activation of the different receptor subtypes with also contrasting effects in promoting or inhibiting angiogenesis . β2-AR is an endogenous mediator of angiogenesis in vivo, as demonstrated in a rat model of neovascularization and adrenergic activation attributable to hind-limb ischemia and later confirmed by the defective angiogenesis in mice knockout for β2-AR [11, 12, 84]. Mechanistic studies show that β2-ARs control specific phenomena involved in angiogenesis such as EC proliferation and stimulating proapoptotic and antiapoptotic pathways. Indeed, β2-AR exerts a positive effect on EC ERK/MAPK activation by at least two mechanisms: first, stimulation of endothelial β-ARs directly activates ERKs; second, β2-AR stimulation can induce the release of VEGF, which can also activate ERKs [11, 85].
The α1A– and α1B-AR subtypes, but not the α1D subtype, are expressed in cultured rat aorta ECs. The activation of these α1-ARs in ECs provide a negative regulation of angiogenesis [12]. Indeed, pharmacological antagonism of α1-AR in ECs from Wistar-Kyoto rats by doxazosin enhanced, while stimulation of these ARs with phenylephrine, inhibited endothelial mechanisms of angiogenesis such as cell proliferation and DNA synthesis, ERK and retinoblastoma protein (Rb) phosphorylation, cell migration, and tubule formation [12]. A similar phenotype can be observed in vivo, since an increased α1-AR density in the ischemic hind limb, compared to nonischemic hind limb, suggested an enhanced α1-AR tone in the ischemic tissue. Treatment with doxazosin did not alter systemic blood pressure but enhanced neoangiogenesis in the ischemic hind limb [12].
ECs and CA Synthesis and Release
ECs are capable of synthesizing and releasing a variety of substances that may exert autocrine, paracrine, or endocrine effects and their function is fine-tuned by several hormones, such as CAs . CAs cause vasoconstriction via α1– and α2-ARs and vasodilation via β2-ARs in vascular smooth muscle [86]. They are synthesized from the amino acid precursor L-tyrosine. Tyrosine hydroxylase (TH) is the first rate-limiting enzyme in CA synthesis that catalyzes the conversion of tyrosine to L-dihydroxyphenylalanine. The latter is converted to dopamine by L-DOPA decarboxylase (DDC). In turn, dopamine is converted to norepinephrine (NE) by dopamine β-hydroxylase (DBH), and NE is converted to epinephrine (Epi) by phenylethanolamine-N-methyl-transferase (PNMT) [87]. Once released, CAs are quickly inactivated by two enzymes that are responsible for their catabolism, catechol-O-methyl-transferase (COMT) and monoamine oxidase (MAO) [87]. MAO catalyzes the oxidative deamination of amines, and COMT methylates the meta-hydroxyl group of CAs.
Initially, circulating CAs were thought to be produced exclusively by the adrenal medulla [88]; over the past few years, however, a growing list of peripheral cell types, such as macrophages, lymphocytes, and even cardiac cells, have been shown to be able to synthesize and release CAs on their own [89–94]. Macrophages, for instance, not only respond to CAs but can also produce them, essentially operating on an autocrine feedback loop in which CAs produced and released by the macrophage activate ARs to regulate interleukin (IL)-1β production, which has a key role in inflammatory responses [95]. ECs have also been added to this list. ECs are capable of synthesizing CAs, because they have the complete intracellular machinery for the generation and release of NE and Epi ([96]. In vitro, ECs express all of the enzymes involved in the synthesis of CAs under normal conditions, and this expression is enhanced in response to specific stimuli such as hypoxia. The molecular mechanism that regulates this phenomenon during hypoxia involves activation of PKA/CREB (cAMP response element-binding protein) signaling, which is known to regulate most important endothelial functions, such as eNOS gene transcriptional activation [97]. Indeed, overexpression of both CREB and PKA enhances DBH and PNMT gene expression and CA release, whereas H89, an inhibitor of PKA, exerts the opposite effect, establishing the role of the PKA/CREB pathway in the regulation of CA release in ECs (Fig. 3.2). These results are corroborated by data from an in vivo model of chronic ischemia, where it is demonstrated that TH, DDC, DBH, and PNMT are expressed in the endothelium of the femoral artery after ischemia.
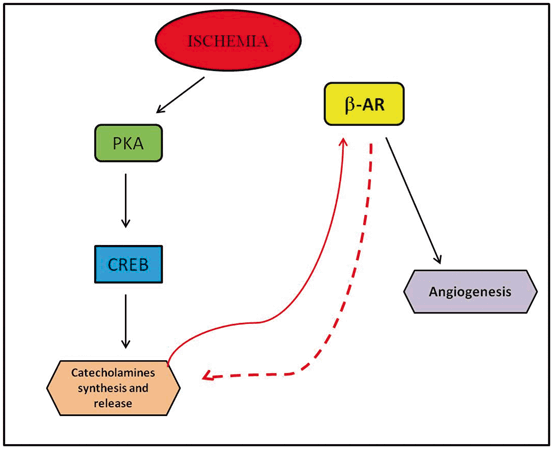
Fig. 3.2
Schematic representation of the regulation of CA synthesis and release in ECs during chronic ischemia. See text for details and definitions of molecular acronyms. PKA protein kinase A, CREB cAMP response element-binding protein, β 2 -AR β2adrenergic receptors
Pathophysiological Implications
Regulation of Vascular Tone and Endothelial Dysfunction
Under physiological conditions, vascular tone is tightly regulated by the adrenergic system, resulting from a balance between α1-AR-mediated vasoconstriction and β2-AR-mediated vasodilatation. Abundant evidence indicates that the defective β-AR-dependent vasodilation during pathological conditions, such as hypertension , is related to the reduced NO production and release, part of a complex state of impaired endothelial function also known as endothelial dysfunction . The link between endothelial dysfunction and vascular diseases is well established [98]. It is known, for instance, that impairment of endothelial function precedes atherosclerosis [7, 9, 16].
As described above, β2-AR activation at the endothelial level induces the production and release of NO, causing vasodilation [10]. In the hypertensive state, this equilibrium is shifted toward increased vasoconstriction, likely because of a defective vasodilatation in response to β2-AR stimulation. Indeed, β-AR agonist administration in the human brachial artery induces vasodilatation and, interestingly, this response is attenuated in hypertensive patients [25, 99]. The role of β2-AR in the vasculature appears to be so critical that genetic variants of this receptor, causing excessive desensitization, may lead to reduced vasodilation [100, 101] and may also promote the occurrence of atherosclerosis [102, 103]. The importance of such a strong molecular interconnection between the adrenergic system and NO release has been also recently demonstrated in several studies with nebivolol conducted in animals and in humans. Nebivolol is a third-generation β-blocker used in the treatment of hypertension which induces vasodilation by increasing NO production. Nebivolol has a distinctive profile among β-blockers, with the greatest selectivity for cardiac β1-ARs and the highest β1 (over β2) selectivity compared with other β-blockers, and no effect on α-receptors. Its ability to enhance NO release has been observed in the human forearm where its vasodilatative effects were blunted by the eNOS inhibitor, L-NG-nitroarginine methyl ester (L-NAME) [104]. Moreover, when compared to atenolol (another β-blocker), nebivolol showed ability to reverse endothelial dysfunction in hypertensive patients [105]. In addition, nebivolol exerts systemic antioxidative properties and this effect is purported to be an additional factor for increasing NO bioavailability. For example, nebivolol and atenolol similarly reduce blood pressure in hypertensive patients, but oxidative stress markers, such as low-density lipoprotein (LDL) hydroperoxides, 8-isoprostanes, and ox-LDL, were significantly improved only with nebivolol. The mechanism by which nebivolol acts on NO bioavailability is still unclear, with some experimental evidence suggesting activation of β3-AR [106–108].
GRK2 and Endothelial Dysfunction
As mentioned above, GRKs play significant roles in adrenergic regulation of endothelial function and their influence is particularly evident in the setting of pathological conditions like hypertension and diabetes . For example, GRK2 participates in the determination of animal model of portal hypertension , relying on its physical interaction with Akt [66]. Since Akt is able to activate eNOS, thereby mediating vasodilation, the GRK2-mediated inhibition of Akt has been shown to shift the vascular tone equilibrium toward constriction, in the setting of endothelial dysfunction, due to decreased eNOS activity [66, 109]. Notably, endothelial modulation of the contractile state of vascular smooth muscle has been shown to be impaired in atherosclerosis and in several conditions known to be associated with the premature development of atherosclerosis. Two different reports, both showing that reducing GRK2 activity can reduce the endothelial dysfunction observed in the hypertensive vasculature, support the role of GRK2 in the pathogenesis of arterial hypertension . The first study demonstrated that, in the aged rat, a model of impaired β-AR signaling, chronic exercise may lead to decreased GRK activity in the vasculature, in particular in the endothelium, and that this reduction mirrors an ameliorated vasodilatation [110]. The other report showed the preventive effect of a specific GRK2 inhibitor on vascular dysfunction in diabetic mice, ameliorating Akt/eNOS impairment and improving the translocation of β-arrestin2 to the plasma membrane [111]. Consistent with these data, acute inhibition of GRK2 using the nonselective inhibitor heparin led to diminished desensitization of isoproterenol-dependent vasodilation in normotensive subjects [25] . Interestingly, heparin administration in hypertensive patients was able to restore the impaired β-AR vasodilation, ameliorating local resistance [25]. Remarkably, the correlation between GRK2 levels and hypertension is also present in other conditions characterized by increased blood pressure, including the aforementioned portal hypertension [66] and preeclampsia [112]. In gestational hypertension, the increase in GRK2 in the fetal–placental vasculature seems to be compensatory rather than causative of increased blood pressure , in order to balance the excessive vascular tension, since the lack of the protective effect of elevated GRK2 expression negatively affected the outcome of the hypertensive state [66]. Therefore, if increased levels of GRK2 are detrimental for EC function, their excessive reduction is also deleterious for cell survival. GRK2 is needed for embryonic development [113] and, in adult life, cardiac-specific GRK2 deletion alters the cardiac hypertrophic response to chronic β-AR stimulation, leading to an eccentric dilatation of the heart similar to that observed in intermediate–advanced phases of heart failure [114, 115]. This kinase appears also fundamental for EC integrity during adult life. The absence of GRK2 in ECs induces cytokine production and macrophage migration into the vascular media, where they release metalloproteinases (MMPs) [7, 116], such as MMP-2 and MMP-9, corrupting the elastic and the connecting fibers that extend through fenestration of the parallel elastic lamellae . These structures are pivotal in maintaining integrity and mechanical properties of the aortic wall [117] and their disruptions are implicated in disease processes, such as atherosclerosis [118], aneurysm formation [119], and in aging [120]. Early atherescloretic lesions can be indeed observed in mice with specific endothelial deletion of GRK2 which also show defective angiogenesis [7, 121]. It is likely that either elevated or reduced levels of GRK2 affect cellular function and survival and this may appear contradictive to the notion of GRK2 inhibition or downregulation as a therapeutic strategy. A reconciling hypothesis might be that GRK2 interacts with and regulates several substrates through its kinase activity, as well as its regulator of G protein signaling (RGS) and plekstrin homology (PH) domains, rendering it capable of multiple regulatory roles inside the cell [122–124]. Moreover, the most effective therapeutic strategy aiming at counteracting GRK2-mediated receptor desensitization at the plasma membrane level has used the carboxyl-terminal portion of GRK2 (β-ARKct), whose mechanisms in animal models of cardiovascular disease are not completely understood. Β-ARKct represents the carboxyl-terminal portion of GRK2, which retains the ability to localize at plasma membrane–residing free Gβγ subunits through its PH domain, but lacks the kinase activity [125, 126]. Β-ARKct thus displaces GRK2 from plasma membranes but does not inhibit its kinase activity; thus, GRK2 is theoretically free to move into other cellular compartments like mitochondria where it accomplishes protective roles as promotion of mitogenesis and cellular adenosine triphosphate (ATP) content preservation. Therefore, any potential therapy involving tight modulation of GRK2 activity/levels must take into account the specific role played by GRK2 in the various subcellular compartments .
Angiogenesis in Vascular Diseases and Tumors: Role of the Adrenergic System
Angiogenesis is considered an important feature of a viable endothelium. Its mechanism entails specific, composite, and coordinated sequences [82] of several cellular and molecular processes, intimately regulated by the ECs [127]. Proliferation, cell migration, and tubule formation by ECs represent the first steps in angiogenesis, leading to the sprouting of immature sinusoids around which a more complex capillary will develop [128]. The connection between angiogenesis and ECs is so close that angiogenesis is now considered to be an aspect of endothelial function and several models of endothelial dysfunction show impaired angiogenesis. As described above, the adrenergic system is pivotal in promoting angiogenesis and this is evident in pathological conditions as chronic ischemia where endogenous CA can activate both endothelial α1– and β2-ARs with opposite effects on EC proliferation, migration, and survival. Apart from the potential clinical and therapeutic applications in cardiovascular disease, in particular during chronic ischemia, it is not surprising that these discoveries can be applied also to oncology. The connection between the adrenergic system and tumor progression has been evidenced in stress conditions where the stress derived from social isolation was found to elevate the tumor NE levels in ovarian cancer patients, and its level was correlated with tumor grades and stages [129]. In particular, pancreatic, breast, ovarian, and colorectal cancers have been extensively investigated about the effects of β-ARs in preclinical and clinical settings [130–132]. A study from Thaker and colleagues revealed that chronic stress could elevate tumor NE levels in an orthotropic ovarian cancer in a mouse model and obviously increased tumor burden and aggressiveness of tumor growth [133]. Propranolol, a nonselective β-AR antagonist, completely abolished the effects of chronic stress on tumor growth. In contrast, the β2-AR-selective agonist terbutaline produced a similar increase in tumor weight just like under chronic stress. Even though the molecular mechanisms are not completely understood, studies in prostate and breast cancer cells also demonstrated that Epi stimulation reduced the sensitivity of cancer cells to apoptosis through β2-ARs/PKA/inactivation of BAD (proapoptotic protein BCL2-associated death promoter) [134]. Preclinical models further confirmed that stress hormones like Epi promoted prostate carcinogenesis through inhibition of apoptosis and tumor involution mediated by an Epi/β2-AR/PKA/BAD antiapoptotic signaling pathway [135]. Moreover, Epi and NE can induce proliferation of colorectal cancer cells preferentially through the β2-AR [132, 136]. Apart from effects on cancer cell proliferation, the adrenergic system can intervene also in angiogenesis , which is known to be essential for tumor growth and metastasis. Several cancer models have shown that Epi and NE can upregulate VEGF and induce tumor angiogenesis and aggressive growth [133, 137–139]. Other angiogenic factors, such as IL-6, IL-8, MMP2, and MMP-9, can be elevated by Epi and NE in a variety of cancer cell lines via β-AR signaling [133, 139, 140]. Therefore, the administration of β-AR antagonists like propranolol could completely abrogate the secretion of these factors and their mediated functions, implying that β-blockers have potential therapeutic value for the management of relevant cancers. This hypothesis is supported by in vivo models of proliferative retinopathies which showed a strong inhibitory role against vascular changes exerted by the blockade of specific β-ARs. In particular, β2-AR seems to be mostly involved in these responses, and the β1-/β2-AR blocker propranolol is highly effective at inhibiting both the increase of VEGF expression caused by a hypoxic insult and the consequent neovascular response. These observations have prompted clinical trials in preterm infants with retinopathy, where oral administration of propranolol produced positive results in terms of efficacy, although safety problems were also reported [141].
The Latest: ECs Synthesize and Release CAs
As demonstrated in other cellular systems, ECs are capable of producing and releasing CAs. These cells possess the entire enzymatic machinery for synthesis and release and are also capable of metabolizing NE and Epi because they also express COMT and MAO. In addition, it appears that this autocrine feedback catecholaminergic loop of the ECs is a positive feedback one, i.e., it leads to further enhancement of CA synthesis and secretion, because β2-ARs present in EC membranes can also stimulate CA synthesis via coupling to the classic Gs protein–adenylyl cyclase–cAMP–PKA–CREB signaling pathway [5]. Therefore, the CAs released by the EC can activate the β2-ARs on its surface and further stimulate their own production. The full pathophysiological implications of this have not yet been elucidated; it appears however that this ability of ECs to produce and release their own CAs serves primarily as a homeostatic, self-protective mechanism to defend the cell against vascular injury/stress and ischemia. Indeed, in mice experiencing hind-limb ischemia secondary to surgical removal of their common femoral artery, expression of the CA biosynthetic enzymes is elevated in the femoral artery and capillary endothelium of the afflicted hind limbs, indicating increased endothelial CA production in response to chronic ischemia in vivo [5]. Thus, ECs appear to be equipped with a “custom-made,” ready-to-respond CA synthesis and release apparatus, which enables them to promptly respond to a vascular injury, a stressful insult, or a hypoxic/ischemic emergency by initiating in situ CA-dependent neoangiogenesis [142]. These findings may have important implications for antihypertensive therapy, as well: Given that some of the most useful antihypertensive drugs are β-blockers, and EC-residing β2-ARs appear instrumental in both CA production and CA-driven angiogenesis by the ECs, it follows that nonsubtype selective β-blockers might be associated with impaired endothelial angiogenesis [142]. Indeed, several lines of evidence indicate that this type of β-blockers might actually be deleterious in heart disease treatment [143]. Thus, β1-AR-selective agents, especially those that combine vasodilatory action, might be preferable for antihypertensive therapy, at least from the endothelial function standpoint [142].
Conclusions
In the past years, great advances have been made in the study of AR signaling and function in the endothelium thanks to the development of new technologies. Indeed, transgenic mouse models have significantly improved our understanding of the mechanisms of action of specific drugs in vivo. The ability to induce transgene expression at defined times or in defined tissues is an important tool, as well as the ability to induce or repress the expression of endogenous genes in a developmental or tissue-specific fashion. Indeed, deletion of the genes encoding for AR subtypes has helped identify the specific subtypes which mediate in vivo effects of specific drugs. Thus, the combination of molecular biological, genetic, and pharmacological techniques greatly facilitates our understanding of AR function in vivo, and, in turn, leads to more effective and specific therapeutic treatment in humans. β-ARs, for instance, are already target of therapeutic intervention in many diseases: β-AR stimulation in asthma and obesity or β-AR blockade in hypertension and coronary insufficiency. In conclusion, given the importance of endothelial function in most physiological and pathological conditions, it is clear that the increasing knowledge of the adrenergic system’s function in the endothelium is instrumental for future progress in its translation to clinical applications.
References
1.
Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–18.PubMed
2.
Lusher JM, Salzman PM. Viral safety and inhibitor development associated with factor VIIIC ultra-purified from plasma in hemophiliacs previously unexposed to factor VIIIC concentrates. The monoclate study group. Semin Hematol. 1990;27:1–7.PubMed
5.
Sorriento D, Santulli G, Del Giudice C, Anastasio A, Trimarco B, Iaccarino G. Endothelial cells are able to synthesize and release catecholamines both in vitro and in vivo. Hypertension. 2012;60:129–136.PubMed
6.
Ferro A, Queen LR, Priest RM, Xu B, Ritter JM, Poston L, Ward JP. Activation of nitric oxide synthase by beta 2-adrenoceptors in human umbilical vein endothelium in vitro. Br J Pharmacol. 1999;126:1872–1880.PubMedCentralPubMed
7.
Ciccarelli M, Sorriento D, Franco A, Fusco A, Del Giudice C, Annunziata R, Cipolletta E, Monti MG, Dorn GW 2nd, Trimarco B, Iaccarino G. Endothelial G protein-coupled receptor kinase 2 regulates vascular homeostasis through the control of free radical oxygen species. Arterioscler Thromb Vasc Biol. 2013;33:2415–24.PubMedCentralPubMed
8.
Campbell WB, Gauthier KM. Inducible endothelium-derived hyperpolarizing factor: role of the 15-lipoxygenase-EDHF pathway. J Cardiovasc Pharmacol. 2013;61:176–87.PubMedCentralPubMed
9.
Iaccarino G, Ciccarelli M, Sorriento D, Cipolletta E, Cerullo V, Iovino GL, Paudice A, Elia A, Santulli G, Campanile A, Arcucci O, Pastore L, Salvatore F, Condorelli G, Trimarco B. AKT participates in endothelial dysfunction in hypertension. Circulation. 2004;109:2587–93.PubMed
10.
Iaccarino G, Cipolletta E, Fiorillo A, Annecchiarico M, Ciccarelli M, Cimini V, Koch WJ, Trimarco B. Beta(2)-adrenergic receptor gene delivery to the endothelium corrects impaired adrenergic vasorelaxation in hypertension. Circulation. 2002;106:349–355.PubMed
11.
Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, Piscione F, Priante O, Pastore L, Chiariello M, Salvatore F, Koch WJ, Trimarco B. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res. 2005;97:1182–89.PubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


