Fig. 18.1
Workflow of Sanger sequencing and two next-generation sequencing platforms. (a) Sanger sequencing. (b) Genome Sequencer FLX from Roche/454. (c) Genome Analyzer from Illumina. dNTP deoxynucleotide, ddNTP dideoxynucleotides, NGS next-generation sequencing, nts nucleotides, Pol DNA polymerase (Figure adapted from Etheridge [56], Shendure and Ji [52], and Mardis [57])
A variety of methods also have been developed for the detection of chromosomal abnormalities. Giemsa staining is a simple and rapid technique for conventional karyotyping and can identify many chromosomal changes including balanced chromosomal aberrations [58]. A higher resolution from tens of kilobases up to several megabases is offered by fluorescence in situ hybridization (FISH), which uses fluorescently labeled probes that hybridize to their complementary chromosomal sequences [58]. As an alternative to these microscopy-based methods, multiplex ligation-dependent probe amplification (MLPA) can be applied, which is based on a multiplexed PCR and can detect copy number changes of up to 50 different loci in parallel [58].
New possibilities for the analysis of genetic variations were provided by microarray-based genotyping, which offers high-resolution genome-wide variation detection and is based on the hybridization of a DNA sample to oligonucleotide probes that have been immobilized on a glass or silicon surface [59]. Array comparative genomic hybridization (array-CGH) is used to identify chromosomal aberrations by comparing a DNA sample to a reference sample. Moreover, DNA microarrays enable the analysis of disease-specific or even genome-wide SNP panels (SNP arrays) [58]. Thus, they allow the detection of known disease-causing mutations in individual patients or the identification of novel associations between SNPs and complex traits in genome-wide association studies (GWAS) [60].
18.4.2 Next-Generation Sequencing
The development of novel high-throughput sequencing technologies has revolutionized biomedical research. These next-generation sequencing (NGS) technologies, first introduced in 2005 [61, 62], have evolved rapidly, and the costs have been reduced from $1000 per megabase to less than $0.1 in 2014 [63]. Thus, it is much more cost efficient than Sanger sequencing ($500 per megabase) and allows a higher degree of parallelization [52]. In contrast to microarrays, NGS is not dependent on DNA hybridization to preselected probes, enabling the identification of novel variations at a single-base resolution without a priori sequence information.
Different NGS platforms have been established, and the companies Roche/454 (Fig. 18.1b), Illumina (Fig. 18.1c), and Life Technologies have set the standard for high-throughput sequencing [64]. Although their systems vary in their chemistry, they are all based on the principle of cyclic-array sequencing. Here, a dense array of DNA features is iteratively enzymatically sequenced combined with imaging-based data collection [52]. In general, a sequencing run generates reads that randomly cover the genome [65]. The coverage describes the average number of times a single base is read during a sequencing run. A higher number of sequence reads result in greater sequencing depth and thus, in higher sequence confidence. For example, within the 1000 Genomes Project, the coverage ranges from low (2–6×) for whole genome sequencing to high (50–100×) for exome sequencing [66].
For the sequencing of genomic DNA, three basic approaches are available [64]. Whole-genome sequencing allows the determination of all genomic variations but is relatively cost intensive. Here, useful alternatives are provided by whole exome and targeted re-sequencing approaches, which require sequence enrichment technologies such as array-based sequence capturing. Whole-exome sequencing enables the sequencing of almost all protein-coding regions (optionally including untranslated regions or long non-coding RNAs), often combined with a high coverage. When knowledge about possible candidate regions (e.g., genes, promoters, and enhancers) and disease pathways is already available, the targeted re-sequencing of these regions is a promising option. The selection of genomic targets for re-sequencing can be based on data from previous projects like sequencing analyses, GWAS, animal models, as well as publicly available databases [64]. Moreover, disease-specific Web resources like the CHDWiki [67] and the Cardiovascular Gene Annotation Initiative, which has annotated more than 4000 cardiovascular-associated proteins [68], provide useful information for candidate gene selection.
Several large cohorts of CHD patients already are under investigation by NGS [64]. For example, the Congenital Heart Disease Genetic Network Study established by the Pediatric Cardiac Genomics Consortium enrolled more than 3700 patients with a diverse range of CHD [69], and so far, whole-exome sequencing data for a subset of 362 patients and their parents is available [70]. Having a broader focus on undiagnosed children with developmental disorders, the Deciphering Developmental Disorders (DDD) study headed by the Wellcome Trust Sanger Institute aims to recruit 12,000 patients and their parents [71]. Recently, exome sequencing and array-CGH were performed for 1113 children and their parents, with CHD occurring in 11 % of the patients [72, 73].
18.4.3 Transcriptome and Epigenome Analysis
Both NGS and array-based technologies are extensively used for transcriptome and epigenome analysis. In addition, quantitative real-time PCR is a useful low- to medium-throughput application. The study of gene expression has been revolutionized by RNA sequencing (RNA-seq), which enables the discovery, profiling, and quantification of RNA transcripts across the entire transcriptome without prior knowledge about the probed sequences. Applications of RNA-seq comprise total RNA-seq (coding and non-coding RNA above a certain size), mRNA-seq (including mRNAs and long non-coding RNAs with a poly-A tail), and small RNA-seq (including microRNAs and other small non-coding RNAs). Novel applications of RNA-seq include de novo transcriptome assembly [74], single-cell transcriptomics [75], and tomography sequencing to determine spatially resolved transcription profiles in whole embryos or isolated organs [76].
A powerful technique for the genome-wide identification of protein–DNA interactions such as transcription factor binding sites or chromatin histone marks is chromatin immunoprecipitation (ChIP). In ChIP, the protein of interest is cross-linked to the DNA, either in cultured cells or in tissue samples. After cross-linking, the chromatin is sheared and an antibody is used to enrich for DNA fragments bound to the protein. Immunoprecipitation and reverse cross-linking isolate the DNA enriched in the binding sites, and finally, the enriched DNA fragments can further be analyzed by hybridization to microarrays (ChIP-chip) or NGS (ChIP-seq) [77, 78] (Fig. 18.2). If candidate target genes or potential sites are available, ChIP-qPCR represents an alternative strategy. To investigate the co-localization of proteins on the DNA, ChIP-reChIP (sequential ChIP) has been developed using two independent rounds of immunoprecipitation [80]. An alternative method used to map protein–genome interactions is DamID, which does not require the use of antibodies. This technique is based on the fusion of the protein of interest to Escherichia coli DNA adenine methyltransferase (dam) and the resulting methylation of adenines in DNA surrounding the native binding sites of the dam fusion partner. In most eukaryotes, adenine methylation does not occur endogenously. Thus, it provides a unique tag to mark protein interaction sites, which can further be identified by array hybridization or NGS [81].
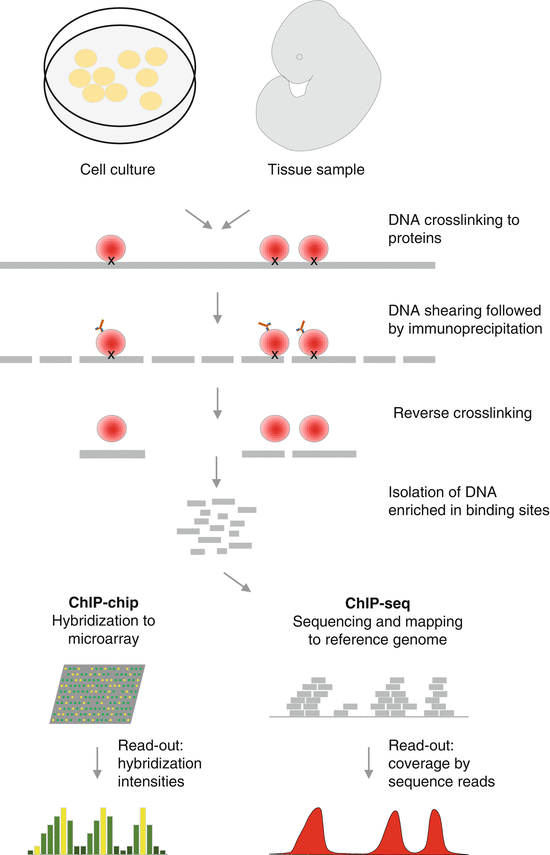
Fig. 18.2
Schematic representation of a chromatin immunoprecipitation (ChIP) experiment followed by microarray detection (ChIP-chip) or next-generation sequencing (ChIP-seq) (Figure adapted from Visel et al. [79])
In addition to histone modifications, DNA methylation occurring on cytosine residues in the context of CpG dinucleotides is also an important epigenetic mark. Altered DNA methylation has been shown to play a role in various diseases, including CHD [82]. Three methods are commonly used to detect genome-wide DNA methylation levels. Two techniques are based on the isolation of methylated DNA fragments by methylated DNA immunoprecipitation (MeDIP) or methyl-CpG binding domain-based (MBD) proteins. Subsequently, the enriched DNA fragments can be detected by arrays or NGS [83]. The third technique applies the treatment of DNA with sodium bisulfite, which converts all non-methylated cytosines to uracil. These will finally be detected as thymine residues, analogous to a C to T SNP, by, for example, pyrosequencing [84] or NGS.
Several techniques are available to assess chromatin structure and regulatory interactions. Chromatin that has lost its condensed structure is sensitive to cleavage by the DNase I enzyme (DNase I hypersensitive sites). Thus, the enzymatic degradation of DNA can be used to identify regions of open chromatin, representing cis-regulatory elements including promoters, enhancers, insulators, and silencers [85]. An alternative method to DNase-seq is the assay of transposase-accessible chromatin (ATAC-seq), which uses an engineered Tn5 transposase to cleave DNA in open chromatin and to integrate primer DNA sequences into the cleaved genomic DNA. Furthermore, a commonly used method to identify the exact positions of nucleosomes is the treatment with micrococcal nuclease (MNase), an endo-exonuclease that processively digests DNA until it is blocked, for example, by a nucleosome [86]. To study interactions between regulatory elements, including long-range interactions between different chromosomes, the chromosome conformation capture (3C) and various derivatives (4C, 5C, and Hi-C) have been developed. They are all based on the cross-linking of interacting DNA fragments and their subsequent restriction digest [87]. Using an additional ChIP step, chromatin analysis by paired-end tag sequencing (ChIA-PET) allows the identification of long-range interactions mediated by target proteins of interest [88].
The interaction of RNAs and proteins is also an important layer for the co-transcriptional and posttranscriptional regulation of gene expression. Genome-wide protein–RNA interaction can be identified based on ultraviolet cross-linking and immunoprecipitation (CLIP). To reach a base-pair resolution, this method was further developed to photoactivatable ribonucleoside-enhanced CLIP (PAR-CLIP), which relies on the incorporation of photoactivatable nucleotide analogues into the RNA. Here, reverse transcription results in a T to C base transition at the cross-link site, detectable as SNPs in the subsequent NGS analysis. However, the need to incorporate photoactivatable nucleotides restricts PAR-CLIP to cultured cells [89]. Finally, a highly sensitive method for the general profiling of RNA-induced silencing complexes (RISC) and individual microRNA target identification is RISC-seq [90].
18.4.4 Proteome and Metabolome Analysis
The quantitative and qualitative large-scale study of proteins (proteomics) and small-molecule metabolites such as alcohols, amino acids, and nucleotides (metabolomics) has undergone great developments over recent years. However, these new technologies have only begun to be applied in CHD research [91, 92], where they have the potential to boost our knowledge of molecular mechanisms underlying heart disease from the pharmacological viewpoint and to enable the discovery of novel biomarkers.
The core technologies for both proteome and metabolome studies are mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy. Most approaches are based on the analysis of peptides, which are frequently generated by enzymatic digestion of proteins. A key step for MS analysis is the selection and enrichment of the proteins/peptides of interest, which can be achieved by subcellular fractioning (e.g., membrane enrichment, nucleus precipitation, or mitochondria separation), by co-immunoprecipitation (e.g., for a protein and its interaction partners), or by enrichment for proteins with particular modifications (e.g., phosphorylation). Furthermore, the development of stable isotope labeling enabled the generation of relative quantitative information [93]. An important technique that can be applied to cell culture studies and more recently, also to studying mouse and drosophila models [94, 95] is stable isotope labeling by amino acids in cell culture (SILAC). Here, two cell populations are cultured in the presence of heavy or light amino acid (e.g., lysine or arginine, respectively) and are further combined for MS analysis [96]. In addition to the metabolic labeling used in SILAC, other methods have been established, including chemical (ICAT and iTRAQ) [97, 98] or enzymatic labeling (18O) [99].
A common method in metabolomics is NMR, which in contrast to MS does not require analyte separation and allows the recovery of the sample for further analyses. It can provide detailed information on the molecular structure of compounds found in complex mixtures like biofluids as well as cell and tissue extracts. NMR offers a high analytical reproducibility and easy sample preparation but is relatively insensitive in comparison to MS [100].
Methods suitable for the high-throughput analysis of protein–protein interactions are the yeast-two-hybrid (Y2H) and the mammalian-two-hybrid (M2H) systems. Both are based on the expression of the two proteins of interest, one fused to the DNA-binding domain and the other to the transactivation domain of a transcription factor, typically Gal4. The binding of the two proteins leads to the complementation of the TF, which activates the expression of a reporter gene (e.g., LacZ). For example, Y2H experiments have been used to identify a large and highly connected network comprising over 3000 interactions between 1705 human proteins [101]. Moreover, a M2H study provided a map of physical interactions within 762 human and 877 mouse DNA-binding transcription factors [102]. In addition to the two-hybrid systems, peptide microarrays have been employed to study protein–protein interactions [103]. However, they have been implemented much slower than DNA arrays due to technical challenges including the high-throughput and economic synthesis of peptides.
An overview of the various molecular biological techniques to study the different regulatory layers that control the gene and protein expression is given in Fig 18.3.
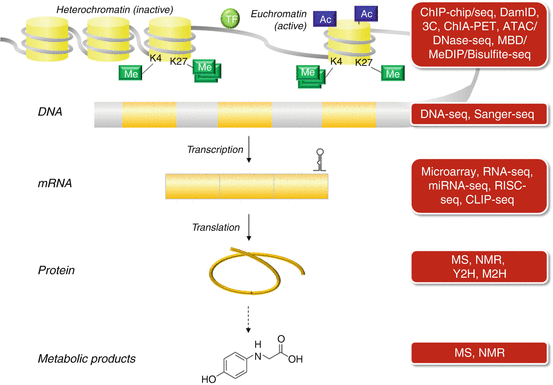
Fig. 18.3
Overview of various molecular biological techniques to study the different regulatory layers controlling gene and protein expression (Figure adapted from Lara-Pezzi et al. [104])
Conclusion
In this chapter, we described various model systems and biotechniques to study the different regulatory levels affecting congenital heart defects. In particular, the application of NGS techniques has revolutionized biomedical research and is still rapidly developing, enabling its application to a wide range of scientific questions. Thus, these high-throughput techniques will enhance our understanding of CHD and will hopefully accelerate the development of novel therapeutic and preventive strategies.
Acknowledgements
This work was supported by the European Community’s Seventh Framework Programme contract (“CardioNeT”) grant 289600 to S.R.S and the German Research Foundation (Heisenberg professorship and grant 574157 to S.R.S.). This work was also supported by the Berlin Institute of Health (BIH-CRG2-ConDi to S.R.S.).
References
1.
2.
3.
4.
Bodmer R (1993) The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development 118:719–729PubMed
5.
Schoenebeck JJ, Yelon D (2007) Illuminating cardiac development: advances in imaging add new dimensions to the utility of zebrafish genetics. Semin Cell Dev Biol 18:27–35PubMedCentralPubMedCrossRef
6.
Molina G, Vogt A, Bakan A et al (2009) Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat Chem Biol 5:680–687PubMedCentralPubMedCrossRef
7.
Major RJ, Poss KD (2007) Zebrafish heart regeneration as a model for cardiac tissue repair. Drug Discov Today Dis Models 4:219–225PubMedCentralPubMedCrossRef
8.
Warkman AS, Krieg PA (2007) Xenopus as a model system for vertebrate heart development. Semin Cell Dev Biol 18:46–53PubMedCentralPubMedCrossRef
9.
Kain KH, Miller JWI, Jones-Paris CR et al (2014) The chick embryo as an expanding experimental model for cancer and cardiovascular research. Dev Dyn 243:216–228PubMedCentralPubMedCrossRef
10.
11.
12.
Bradley A, Anastassiadis K, Ayadi A et al (2012) The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome 23:580–586PubMedCentralPubMedCrossRef
13.
Andersen TA, Troelsen Kde LL, Larsen LA (2014) Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci 71:1327–1352
14.
Winston JB, Erlich JM, Green CA et al (2010) Heterogeneity of genetic modifiers ensures normal cardiac development. Circulation 121:1313–1321PubMedCentralPubMedCrossRef
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


