Fig. 18.1
Giant cell arteritis. (a) Low-power photomicrograph of aortitis due to giant cell arteritis with large-vessel involvement. In the middle of the aortic media (arrows) is a band of acellular collapsed elastic tissue surrounded by an inflammatory infiltrate. (b) Low-power photomicrograph of a temporal artery biopsy involved by giant cell arteritis. There is transmural inflammation, resulting in luminal narrowing. (c) Higher-power photomicrograph of the aortic wall seen in (a). The inflammation is rich in histiocytes and lymphocytes and includes multinucleated giant cells (arrows). (d) Higher-power photomicrograph of the temporal artery wall seen in (b). The inflammation is mixed but rich in histiocytes and lymphocytes and includes multinucleated giant cells (arrows)
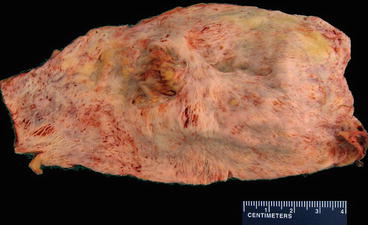
Fig. 18.2
Giant cell arteritis. An opened surgical resection specimen from a patient with ascending aortic aneurysm due to giant cell arteritis with large-vessel involvement. Note the linear wrinkles that have been described as having a “tree bark” appearance. These result from scar replacement of elastic tissue in the aortic wall
18.2.1.3 Presentation/Diagnosis
GCA is a clinical diagnosis, incorporating both symptoms and laboratory markers of inflammation. The gold standard for diagnosis remains temporal artery biopsy. Patients typically present with symptoms such as unilateral headache, fatigue, fever, jaw pain, myalgias, and diaphoresis [4]. High morbidity may occur from permanent vision loss due to inflammation of the ophthalmic artery. Additionally, there is a markedly increased risk of ascending aortic aneurysms and aortic rupture in patients diagnosed with GCA [5].
18.2.1.4 Treatment
Treatment consists of a long course of high-dose corticosteroids. Treatment is often started empirically, before biopsy results, as vision may be at risk (retinal artery occlusion) if treatment is delayed [6].
18.2.2 Takayasu Arteritis
Takayasu arteritis is a chronic granulomatous inflammatory disease of unknown etiology that affects the aorta and its primary branches, but not the small carotid artery branches. It is also distinguished from GCA in that it almost exclusively affects younger women, often of Asian ancestry [7].
18.2.2.1 Epidemiology
Takayasu arteritis has an estimated incidence of 1.2/1,000,000–2.6/1,000,000 individuals annually. It is a disease of adolescents and young adults. While the diagnostic age criterion is <50 years of age [2], the great majority have signs or symptoms decades before age 50.
18.2.2.2 Pathology/Pathophysiology
Like GCA, Takayasu arteritis is characterized by focal granulomatous inflammation of the large vessels. Unlike GCA, the adventitial inflammation is typically more impressive than medial inflammation (Fig. 18.3). Neutrophils and areas of necrosis may be seen as well. The etiology and pathogenesis are not well understood, although the apparent aggressiveness of this disease suggests a more active T-cell-mediated process with acute inflammation, followed by necrosis, neovascularization, intimal proliferation with smooth muscle integration, and finally giant cell formation in the remodeled vessel walls [7].
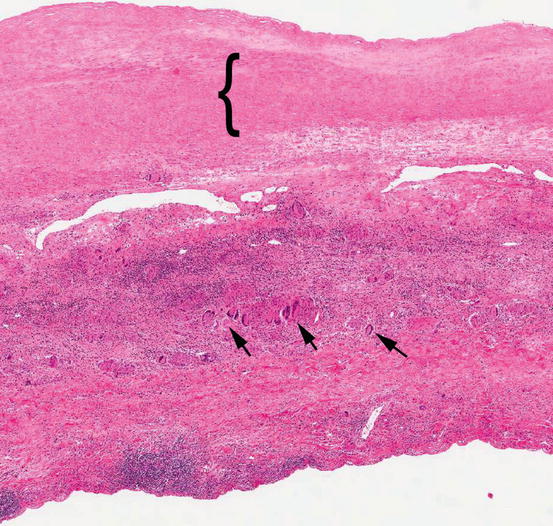
Fig. 18.3
Takayasu arteritis. Low-power photomicrograph of an aortic wall removed from a patient with Takayasu arteritis showing inflammation throughout the wall. The media is demarcated by a ({) symbol. Most of the wall thickness is a result of adventitial inflammation and fibrosis. Multinucleated giant cells (arrows) can be seen even at low magnification
18.2.2.3 Presentation/Diagnosis
Aortitis with ascending aortic aneurysm is the most common presentation. The affected aortic branches (subclavian, carotid, renal, mesenteric) are prone to obstructive stenosis, so Takayasu arteritis is also known as “pulseless disease.” Patients initially present with systemic symptoms such as fevers, chills, malaise, diaphoresis, myalgias, anorexia, and weight loss. Patients often progress to vascular complications such as stenosis, occlusions, and cerebrovascular events. Diagnostic criteria have been delineated by both the American College of Rheumatology and the Ishikawa Criteria.
18.2.2.4 Treatment
Treatment for Takayasu arteritis generally consists of a long course of immunosuppression with corticosteroids and additional immunomodulatory agents depending on disease severity and acuity [8].
18.3 Medium-Vessel Vasculitis
18.3.1 Polyarteritis Nodosa
Polyarteritis nodosa (PAN) is the prototypic medium-vessel vasculitis, showing transmural necrotizing arteritis of medium-sized arteries. It is not associated with antineutrophilic cytoplasmic antibodies (ANCAs). Arterioles, capillaries, venules, and glomeruli are spared. Additionally, the current definition of PAN excludes hepatitis B-associated vasculitis, a specific disease once considered a variant of PAN [1, 9].
18.3.1.1 Epidemiology
PAN is a relatively uncommon disease, occurring in an estimated 4–9/1,000,000 individuals [10]. Previous estimates were much higher prior to reclassification of the disease to exclude hepatitis B-associated vasculitis.
18.3.1.2 Pathology/Pathophysiology
PAN is a systemic, transmural necrotizing vasculitis of the medium-sized vessels. The transmural vessel wall damage leads to pseudoaneurysm (contained rupture) formation in the affected vessels. The size of the pseudoaneurysms is many times greater than the adjacent vessel diameter and they often thrombose, leading to the appearance of nodules, hence “nodosa” (Fig. 18.4). The majority of cases affect the kidneys, gastrointestinal tract, and skin [11]. CNS involvement is rare. Temporal variability (both recent and old lesions present at the time of diagnosis) is a hallmark of PAN. The etiology of PAN is unknown. Innate and humoral immunity likely participate given the rapid and destructive nature of the immune response. A large portion of previous PAN diagnoses were likely hepatitis B-associated vasculitis, confounding much of the previous research on PAN [12].
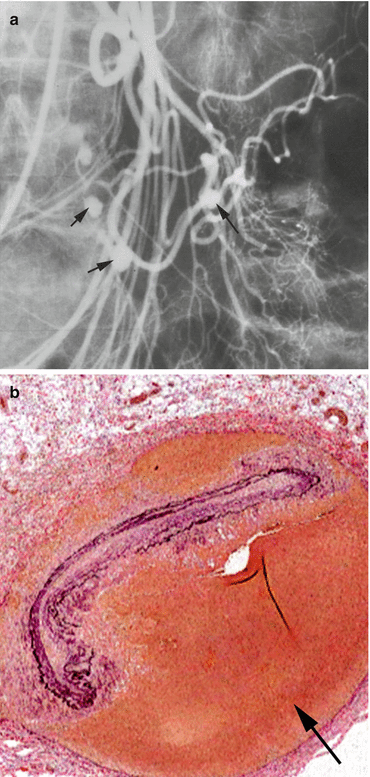
Fig. 18.4
Polyarteritis nodosa. (a) Mesenteric artery angiogram in a patient with polyarteritis nodosa showing multiple nodular pseudoaneurysms (arrows). (b) Low-power photomicrograph of a pseudoaneurysm in polyarteritis nodosa. The collapsed elastic laminae represent the former true lumen; the arrow indicates the aneurysm formed by contained rupture of the native artery
18.3.1.3 Presentation/Diagnosis
18.3.1.4 Treatment
Treatment of PAN involves high-dose corticosteroids with the addition of cyclophosphamide and other cytotoxic agents for more severe cases [14].
18.3.2 Kawasaki Disease
Kawasaki disease (KD) is an arteritis predominantly involving medium and small arteries. It is associated with mucocutaneous lymph node syndrome.
18.3.2.1 Epidemiology
KD is most common in infants and young children although its sequelae are lifelong. KD incidence in the United States is approximately 17–20/100,000 individuals; however, it is significantly more common in East Asians with an incidence of approximately 220/100,000 [15]. In the developed world, as rheumatic heart disease has declined in recent decades, KD has become the leading cause of acquired cardiovascular disease in children [16].
18.3.2.2 Pathology/Pathophysiology
The etiology of KD is still unknown. A growing body of evidence indicates autoimmune reactivity to viral proteins (mimicry) influenced by a genetic predisposition [17]. The most important consequence of KD is acquired cardiovascular disease due to coronary artery vasculitis with pseudoaneurysm formation (Fig. 18.5). The vascular lesions consist of a transmural inflammation with eventual accumulation macrophages and IgA plasma cells [18]. Severe damage to all layers of the arterial wall occurs, weakening the wall and leading to pseudoaneurysm formation in a fashion similar to PAN [19].
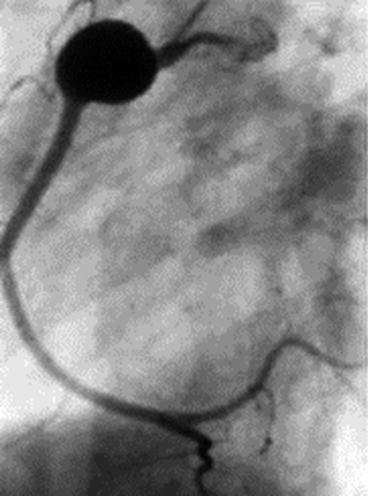
Fig. 18.5
Kawasaki disease. Right coronary artery angiogram showing a pseudoaneurysm in an adult patient who had Kawasaki disease as a child
18.3.2.3 Presentation/Diagnosis
KD is a clinical diagnosis based on these criteria: (1) fever for 5 or more days, (2) bilateral conjunctival congestion, (3) changes in the lips and oral cavity, (4) polymorphous exanthema, (5) changes in peripheral extremities including erythema or desquamation, and (6) acute non-purulent cervical lymphadenopathy [20]. Coronary pseudoaneurysms may not be present in the early phases of illness and coronary arteritis may not be apparent until later on.
18.3.2.4 Treatment
Treatment consists of high-dose intravenous immunoglobulin [21]. Prompt treatment decreases the likelihood of progression to coronary artery abnormalities. A variety of other immunomodulatory treatments have been attempted with varied success. High-dose intravenous immunoglobulin remains the first-line treatment [22].
18.4 Small-Vessel Vasculitis
18.4.1 ANCA Associated
18.4.1.1 Granulomatosis with Polyangiitis (Wegener’s)
Granulomatosis with polyangiitis (GPA) also termed “Wegener’s granulomatosis” is a necrotizing vasculitis of the respiratory tract and small- to medium-sized vessels. Renal involvement is common.
Epidemiology
Pathology/Pathophysiology
Like PAN and KD, the inflammation in GPA and the other small-vessel vasculitis syndromes is acute and necrotizing. Small-vessel vasculitis typically manifests in capillary-sized vessels, particularly in the renal glomerulus and alveolar capillaries of the lung. GPA has three major pathologic features: necrotizing granulomas of the respiratory tract, granulomatous necrotizing vasculitis of small (and medium) vessels, and necrotizing crescentic glomerulonephritis [25] (Fig. 18.6).
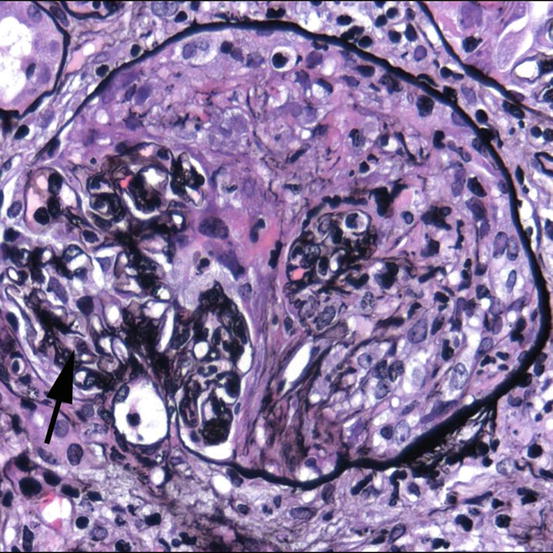
Fig. 18.6
Granulomatosis with polyangiitis. High-power photomicrograph of a glomerulus affected by necrotizing crescentic glomerulonephritis in a patient with positive PR3/c-ANCA. Residual intact glomerular capillaries are seen in the lower left (arrow). The rest of Bowman’s space is occupied by proliferating parietal epithelial cells, leukocytes, and organizing fibrin
Presentation/Diagnosis
Patients often present with respiratory complaints, both lower and upper respiratory tract. This includes nasal septum ulceration, subglottic stenosis, hemoptysis, pneumonias, and eye irritation. The so-called pulmonary-renal syndrome is the combination of these signs and symptoms with glomerulonephritis [24]. GPA is a clinical diagnosis with two or more of the following criteria to be met: nasal or oral inflammation, abnormal chest imaging, hematuria, and granulomatous inflammation on a biopsy specimen [26]. Antibodies against the neutrophil cytoplasmic protein protease 3 (PR3) can be detected in the serum. In mouse neutrophil staining assays, this autoantibody yields a cytoplasmic staining pattern (c-ANCA) (Fig. 18.7).
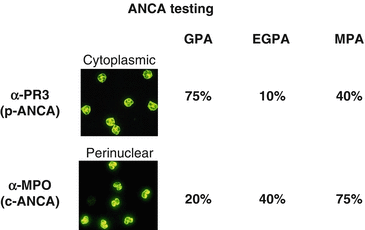
Fig. 18.7
Antineutrophil cytoplasmic antibody testing. (Abbreviations: ANCA antineutrophil cytoplasmic antibody, PR3 proteinase 3, MPO myeloperoxidase, GPA granulomatosis with polyangiitis, EGPA eosinophilic granulomatosis with polyangiitis, MPA microscopic polyangiitis)
Treatment
Treatment for GPA generally consists of a long course of cyclophosphamide and prednisone [27]. GPA portended an extremely high mortality prior to development of effective treatment strategies (mean survival of less than 5 months and a 2-year mortality of 93 % [27, 28]). This is no longer the case, although GPA can have relapsing-remitting nature with up to 50 % recurrence despite aggressive treatment. There is still significant disease morbidity as well [28, 29].
18.4.1.2 Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss Syndrome)
Eosinophilic granulomatosis with polyangiitis (EGPA), formerly Churg-Strauss syndrome, is a necrotizing granulomatous small-vessel vasculitis with extensive eosinophilia that is associated with asthma [1].
Epidemiology
EGPA typically develops in asthmatic patients in their fifth decade of life, and recent data indicate 5-year survival at greater than 90 % with appropriate therapy [30]. Epidemiologic data varies widely regarding EGPA with incidence reports ranging between 0.5 per million and 70 per million individuals [31], depending on the population.
Pathology/Pathophysiology
EGPA is a systemic vasculitis affecting small to medium vessels and is associated with asthma and eosinophilia. Necrotizing vasculitis with eosinophilia and a variable granulomatous component is found on histologic examination of affected tissues (e.g., lung and kidney) [32]. Lesions are often found in the upper airways, heart, skin, and neurovascular bundles as well. Antibodies directed against the neutrophil cytoplasmic protein myeloperoxidase (MPO) can be detected in the serum. In mouse neutrophil staining assays, this autoantibody yields a perinuclear staining pattern (p-ANCA). MPO-ANCA is detected in ~40 % of cases and indicates a worse prognosis [33] when present. The etiology is not well understood, but the association with asthma suggests a common underlying immune mechanism.
Presentation/Diagnosis
Patients almost always have preexisting asthma or develop adult-onset asthma. Patients then begin to experience systemic symptoms such as fevers, malaise, anorexia, and weight loss. Additional symptoms affecting the sinuses and upper respiratory system are frequent [34]. Diagnosis is made when a patient presents with a history of asthma, eosinophilia, and systemic vasculitis [35].
Treatment
Treatment for EGPA consists of a long course of corticosteroids with or without the addition of cyclophosphamide depending upon disease severity [36].
18.4.1.3 Microscopic Polyangiitis
Microscopic polyangiitis (MPA) is an uncommon necrotizing vasculitis affecting small vessels. Like GPA and EGPA, MPA is pauci-immune process. While ANCA may be detectable in serum, no bound antibody or immune complexes are demonstrated by tissue pathology techniques [1]. This is because antibody-mediated neutrophil damage occurs in the peripheral circulation, and the dying neutrophils lodge in the capillary beds (especially renal and pulmonary capillaries) and release their toxic contents. Antibody and immune complexes are absent in the tissue by immunofluorescence techniques at this stage of the disease.
Epidemiology
MPA is a relatively uncommon vasculitis, with prevalence estimates ranging from 2.7 to 94 cases per million individuals in Europe [37, 38]. Typical age of onset is in the fifth decade of life with no sex predilection [39].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


