Sympathetic Nervous System in Heart Failure
G. Michael Felker
Wilson S. Colucci
Howard A. Rockman
The sympathetic nervous system (SNS) plays a central role in cardiovascular physiology by releasing endogenous neurohormones that primarily act on a family of cellular receptors known as 7-transmembrane or heptahelical receptors. Activation of these heptahelical receptors transduces extracellular signals across the cell surface to mediate numerous cellular processes. In the event of an acute decrease in myocardial performance, increased SNS activity serves as a critical compensatory mechanism that supports the cardiovascular system. This is accomplished through a variety of physiology changes, including increasing heart rate, myocardial contractility, and venous return, all of which help to restore and maintain cardiac output. Adequate blood pressure to preserve organ perfusion is further supported by systemic arterial constriction, and intravascular volume is expanded through increased renal retention of sodium and water. While adaptive in the short term, these have deleterious effects on the cardiovascular system when maintained chronically. Vasoconstriction, salt and water retention, increased heart rate, and increased contractility lead to increases in myocardial wall stresses, oxygen consumption, and energetic requirements. Chronically elevated levels of SNS activity may also cause arrhythmias, desensitization of the postsynaptic β-adrenergic receptor pathway, and activation of other neurohumoral systems (e.g., the renin-angiotensin system), which may themselves exert adverse effects. Finally, prolonged exposure to norepinephrine (NE) may contribute to disease progression by acting directly on the biology of the myocardium to modify cellular phenotype and cause myocyte death (1). The balance between the compensatory and the maladaptive effects of SNS activation is central to the understanding of heart failure pathophysiology.
Regulation of Sympathetic Nervous System in Heart Failure
Sympathetic activity, as measured by an increase in circulating plasma NE, is increased in patients with heart failure and is related to both disease severity and prognosis (Fig. 11-1) (2). In addition to elevations in plasma NE, SNS activation in heart failure is demonstrated by increases in the urinary excretion of catecholamines and electrical activity in skeletal muscle (2,3,4,5,6). Of note, plasma NE is elevated even in patients with mild or asymptomatic heart failure in whom there is little or no increase in circulating renin levels (7). While plasma NE levels reflect overall systemic activity, they do not address the level of cardiac sympathetic activity. Direct measurements of cardiac sympathetic nerve activity are not feasible in patients. However, the aorta-to-coronary-sinus gradient of plasma NE is increased in patients with heart failure (8), indicating an increase in the net release of NE from the heart.
Studies using tracer quantities of 3H-NE to measure NE turnover kinetics across the heart have provided additional important information about cardiac sympathetic activity. These techniques have demonstrated that NE spillover is increased in the heart of patients with more severe heart failure (Fig. 11-2) (9,10). Cardiac spillover of NE is increased even in patients with mild symptoms of heart failure, at a time when there is no increase in total body or renal NE spillover (11), indicating that with early heart failure there is selective activation of cardiac sympathetic nerves.
An increase in cardiac spillover may reflect an increase in NE release, a decrease in reuptake, or both (10). In one study (12), cardiac NE spillover and dihydroxyphenylalanine (DOPA) production were increased eightfold and
twofold, respectively, in patients with heart failure, whereas cardiac extraction of NE was unchanged. The outward flux of dihydroxylphenylglycol (DHPG), which reflects neuronal reuptake and intraneuronal metabolism of NE, was similar in normal subjects and patients with heart failure and was reduced to a comparable degree by desipramine, which blocks neuronal reuptake, suggesting that their NE reuptake was not impaired. However, another study found a modest reduction in neuronal reuptake (13), leading to the conclusion that although increased NE release is the major mechanism of NE spillover, reduced reuptake may contribute, particularly in patients with more severe heart failure (10). The mechanism responsible for increased neuronal release of NE in heart failure is not certain but, as discussed below, may involve reflex mechanisms and/or presynaptic facilitation of NE release.
twofold, respectively, in patients with heart failure, whereas cardiac extraction of NE was unchanged. The outward flux of dihydroxylphenylglycol (DHPG), which reflects neuronal reuptake and intraneuronal metabolism of NE, was similar in normal subjects and patients with heart failure and was reduced to a comparable degree by desipramine, which blocks neuronal reuptake, suggesting that their NE reuptake was not impaired. However, another study found a modest reduction in neuronal reuptake (13), leading to the conclusion that although increased NE release is the major mechanism of NE spillover, reduced reuptake may contribute, particularly in patients with more severe heart failure (10). The mechanism responsible for increased neuronal release of NE in heart failure is not certain but, as discussed below, may involve reflex mechanisms and/or presynaptic facilitation of NE release.
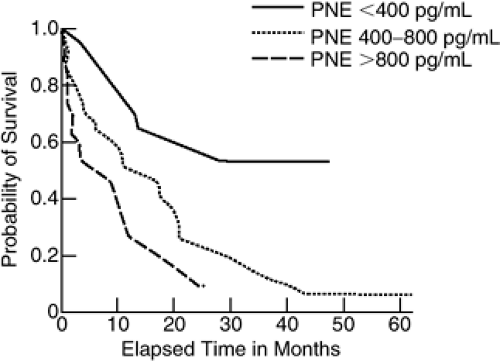 Figure 11-1 Plasma norepinephrine levels in patients with heart failure. Patients with the most elevated levels had the worst prognosis. (From Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–820 , with permission.) |
Blunted Baroreceptor Function in Heart Failure
Baroreceptor function is abnormal in both experimental animals and humans with heart failure (14,15). Increases in mean arterial or pulse pressure, or increased atrial or ventricular filling pressures and distention, or both, normally cause activation of cardiopulmonary and arterial baroreceptors, which inhibit SNS outflow from cardiovascular centers and cause a reciprocal increase in parasympathetic activity. Baroreceptor sensitivity is blunted in animal models and patients with heart failure, as evidenced by the observation that the reflex sympathetic response to reductions in arterial pressure (e.g., caused by nitroprusside infusion, head-up tilt, or lower-body negative pressure) is decreased (14,15,16,17,18,19). Conversely, the reflex withdrawal of sympathetic tone that normally follows the infusion of a vasopressor (e.g., phenylephrine) is attenuated.
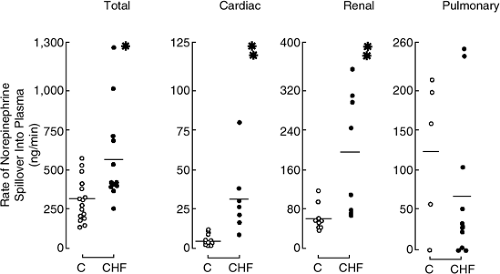 Figure 11-2 Total and regional norepinephrine spillover in patients with congestive heart failure and normal subjects. •, p <0.02; ••, p <0.002. (From Hasking GJ, Esler MD, Jennings GL, et al. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation. 1986;73(4):615–621 , with permission.) |
Baroreceptor sensitivity can be quantified by measuring the relative changes in heart rate (R-R interval) and arterial pressure during the infusion of phenylephrine, which in normal persons causes an approximately 10-millisecond increase in the R-R interval for each 1 mm Hg rise in arterial pressure. In patients with severe heart failure, this slope is markedly reduced to the range of 2 ms/mm Hg (Fig. 11-3). Cardiac transplantation causes normalization of baroreceptor function with a return of the slope to the normal range (20), suggesting that baroreceptor dysfunction is not a primary abnormality but, rather, is secondary to cardiac dysfunction. An important consequence of reduced baroreceptor sensitivity may be that SNS efferent activity is inappropriately elevated relative to the level of cardiovascular performance.
The cellular basis for baroreceptor dysfunction is not known but may involve a number of factors such as mechanical alterations in the environment of the baroreceptor (e.g., vessel wall, myocardium), a resetting of baroreceptor reflex pathways (perhaps at the brainstem level), or alterations in the biochemical function of the baroreceptor, such as altered Na+,K+-ATPase activity (15). In animals, perfusion of isolated baroreceptors with digitalis or low K+ to inhibit Na+,K+-ATPase partially corrects abnormal baroreceptor sensitivity (16), suggesting that increased Na+,K+-ATPase activity may contribute to reduced baroreceptor sensitivity in heart failure. Likewise, in patients with heart failure the acute intravenous administration of a digitalis glycoside normalizes baroreceptor sensitivity, as reflected by decreases in both forearm vascular
resistance and sympathetic nerve activity to skeletal muscle (21).
resistance and sympathetic nerve activity to skeletal muscle (21).
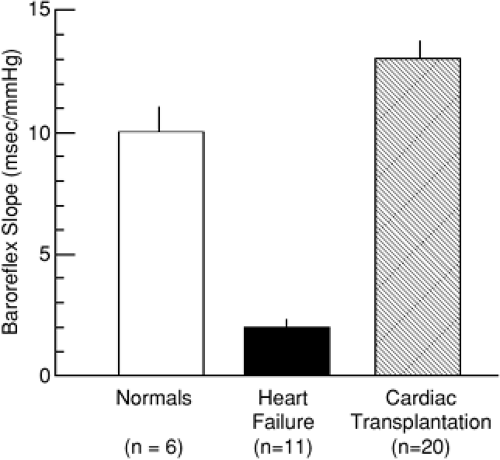 Figure 11-3 Arterial baroreflex slopes in normal persons, patients with congestive heart failure, and patients after cardiac transplantation. The arterial baroreflex slope is determined as the increase in R-R interval (milliseconds) per increase in arterial blood pressure (mm Hg) during administration of a vasoconstrictor such as phenylephrine. The baroreflex slope is typically reduced in patients with congestive heart failure, reflecting reduced sensitivity of arterial baroreceptors. After cardiac transplantation, baroreceptor sensitivity is restored to normal, indicating that reduced baroreceptor sensitivity in heart failure is reversible. (From Hirsch AT, Dzau VJ, Creager MA. Baroreceptor function in congestive heart failure: effect on neurohumoral activation and regional vascular resistance. Circulation. 1987;75(5 Pt 2):IV36–IV48 , with permission.) |
Most studies of reflex regulation in heart failure have focused on arterial baroreceptor function. It has been observed that cardiac NE spillover is increased in proportion to right heart pressures in patients with heart failure (22), leading to the suggestion that increased pulmonary pressures might be a stimulus for increased SNS activity. However, in contrast to arterial baroreceptors, the responsiveness of low-pressure cardiac receptors appears to be relatively preserved in patients with heart failure and would be anticipated to mediate a decrease rather than an increase in sympathetic activity in response to increased filling pressures (23).
Potentiation of Peripheral Norepinephrine Release
Norepinephrine is stored in vesicles in sympathetic nerve endings, where exocytotic release is triggered by a membrane depolarization-induced increase in the intracellular calcium concentration. The amount of NE released in response to a nerve impulse can be modulated by a variety of hormones and substances that act on specific receptors located on the nerve ending (Table 11-1) (24). By this mechanism, stimulation of presynaptic α2-adrenergic receptors decreases NE release, whereas stimulation of β2-adrenergic or angiotensin (25) receptors increases NE release. In humans it can be shown that the nonselective α-adrenergic antagonist phentolamine, which inhibits presynaptic α2-adrenergic receptors, increases NE release in the forearm (26) and heart (27). Conversely, stimulation of β2-adrenergic receptors increases NE spillover from the heart (28).
Table 11-1 Presynaptic Receptors that May Modulate the Release of Norepinephrine | ||||
|---|---|---|---|---|
|
Given the increased levels of angiotensin and NE in heart failure, these observations could have important pathophysiological and therapeutic implications. Converting enzyme inhibitors and angiotensin receptor-blockers may reduce NE release by reducing the availability of angiotensin at presynaptic nerve endings. Conversely, increased NE availability at the nerve ending may facilitate additional NE release. Likewise, there may be release of epinephrine from extraneuronal sources in the hearts of patient with heart failure (29), which may in turn facilitate NE release via stimulation of presynaptic β2-adrenergic receptors.
Neuronal Norepinephrine Stores
Adrenergic nerves take up tyrosine, which is acted on by a series of enzymes (tyrosine hydroxylase, amino acid decarboxylase, and dopamine β-hydroxylase) to yield NE (30). Most of the NE released from the nerve ending is taken up by neuronal and, to a lesser extent, extraneuronal tissues (e.g., endothelial cells, vascular smooth muscle cells). The NE taken up by neurons may be recycled into vesicles for future release or converted to metabolites that are released into the circulation. The NE taken up by extraneuronal cells is likewise converted to metabolites that are released to the interstitial space and may be recovered in the plasma. As discussed previously, a decrease in the reuptake of NE by sympathetic neurons may contribute to increased NE spillover in patients with heart failure (10). Early work in patients with heart failure and in animal models found that the tissue concentration of NE was decreased in myocardium (3,30). Likewise, the release of myocardial NE stores by tyrosine was decreased in patients with heart failure. This depletion of cardiac NE may reflect a defect in NE biosynthesis (30) or a decrease in the neuronal reuptake of NE (13).
Postsynaptic Regulation of the Adrenergic Response
β-adrenergic responsiveness is reduced in patients with heart failure, a phenomenon known as desensitization. In vitro,
myocardium obtained from patients with end-stage heart failure at the time of transplantation exhibits a normal contractile response to calcium but a markedly attenuated response to β-adrenergic agonists (31). In situ, the positive inotropic response to intracoronary infusion of a β-adrenergic agonist (dobutamine) is markedly depressed (Fig. 11-4) (32), as is the chronotropic response to a β-adrenergic agonist (isoproterenol) or exercise (Fig. 11-5) (33). Because heart rate is the predominant determinant of the increase in cardiac output that occurs with exercise, this may be a major way in which β-adrenergic receptor pathway dysfunction impacts the physiology of a patient with heart failure. The degree of attenuation of the inotropic and chronotropic response to β-adrenergic stimulation is inversely related to plasma NE (32,33). In contrast, the isovolumic relaxation response to β-adrenergic receptor stimulation is preserved, even in patients with end-stage heart failure (34). Several mechanisms may contribute to reduced β-adrenergic responsiveness in heart failure (Table 11-2). In the following, we summarize the body of animal and human studies that contribute to the current understanding of regulation of β-adrenergic responsiveness at the receptor level.
myocardium obtained from patients with end-stage heart failure at the time of transplantation exhibits a normal contractile response to calcium but a markedly attenuated response to β-adrenergic agonists (31). In situ, the positive inotropic response to intracoronary infusion of a β-adrenergic agonist (dobutamine) is markedly depressed (Fig. 11-4) (32), as is the chronotropic response to a β-adrenergic agonist (isoproterenol) or exercise (Fig. 11-5) (33). Because heart rate is the predominant determinant of the increase in cardiac output that occurs with exercise, this may be a major way in which β-adrenergic receptor pathway dysfunction impacts the physiology of a patient with heart failure. The degree of attenuation of the inotropic and chronotropic response to β-adrenergic stimulation is inversely related to plasma NE (32,33). In contrast, the isovolumic relaxation response to β-adrenergic receptor stimulation is preserved, even in patients with end-stage heart failure (34). Several mechanisms may contribute to reduced β-adrenergic responsiveness in heart failure (Table 11-2). In the following, we summarize the body of animal and human studies that contribute to the current understanding of regulation of β-adrenergic responsiveness at the receptor level.
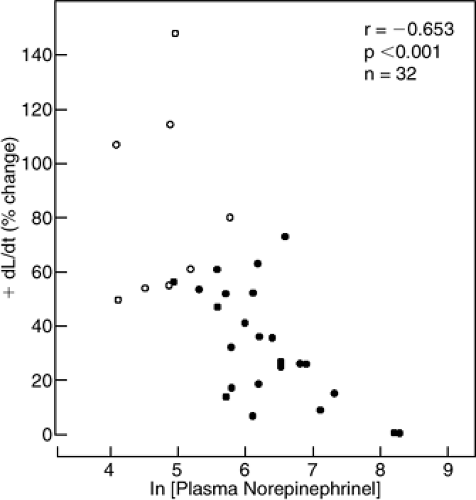 Figure 11-4 The positive inotropic response to an intracoronary infusion of dobutamine, 25 μg/min, in patients with congestive heart failure (solid circles) and persons without congestive heart failure (open circles). The magnitude of positive inotropic response to β-adrenergic receptor stimulation with intracoronary dobutamine is inversely related to the resting plasma NE concentration at the time of study, suggesting that decreased β-adrenergic responsiveness of the heart in patients with heart failure is caused, at least in part, by tonic elevation of sympathetic tone. (From Colucci WS, Denniss AR, Leatherman GF, et al. Intracoronary infusion of dobutamine to patients with and without severe congestive heart failure. Dose-response relationships, correlation with circulating catecholamines, and effect of phosphodiesterase inhibition. J Clin Invest. 1988;81(4):1103–1110 .) |
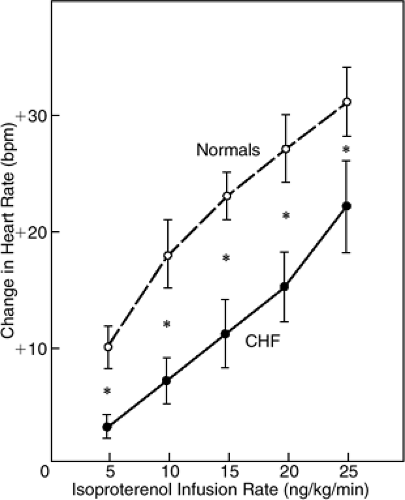 Figure 11-5 Dose-response relationship for isoproterenol-stimulated heart rate increase in normal persons (dashed line) and patients with severe congestive heart failure (solid line). (From Colucci WS, Ribeiro JP, Rocco MB, et al. Impaired chronotropic response to exercise in patients with congestive heart failure. Role of postsynaptic beta-adrenergic desensitization. Circulation 1989;80(2):314–323 , with permission.) |
Biology of Heptahelical Receptors
Adrenergic receptors belong to the large superfamily of receptors known as 7-transmembrane or heptahelical receptors. Two classes of adrenergic receptors (ARs), α and β, have been described and, even though they both respond to norepinephrine and epinephrine, the cellular responses they mediate differ significantly. There are three known subtypes of βARs (β1, β2, and β3), all of which are expressed in the heart (35) wherein, despite the existence of species-related differences (36), β1ARs are the most abundant.
Activation of all types of adrenergic receptors results in G-protein-mediated generation of second messengers and/or activation of ion channels; therefore, these receptors are commonly referred to as G-protein-coupled receptors (GPCRs). By inducing conformational changes in the receptor, agonist stimulation allows interaction with the cognate heterotrimeric G-proteins, promoting dissociation
of G-proteins into Gα and Gβγ subunits and activation. Both Gα subunits and Gβγ dimers amplify and propagate signals intracellularly by activating one or more effector molecules, which in turn activate different signaling pathways by generating second messengers (Fig. 11-6). All known βAR subtypes couple to Gs and activate adenylyl cyclase, resulting in elevated cyclic adenosine monophosphate (cAMP) levels and subsequent activation of protein kinase (PKA) (37). PKA activation is a critical step in the mediation of contractility through phosphorylation of L-type calcium channels and regulation of calcium influx and reuptake (36). In addition to Gs, β2ARs have been shown to couple to Gi both in vitro (38), and in the heart (36,39,40,41).
of G-proteins into Gα and Gβγ subunits and activation. Both Gα subunits and Gβγ dimers amplify and propagate signals intracellularly by activating one or more effector molecules, which in turn activate different signaling pathways by generating second messengers (Fig. 11-6). All known βAR subtypes couple to Gs and activate adenylyl cyclase, resulting in elevated cyclic adenosine monophosphate (cAMP) levels and subsequent activation of protein kinase (PKA) (37). PKA activation is a critical step in the mediation of contractility through phosphorylation of L-type calcium channels and regulation of calcium influx and reuptake (36). In addition to Gs, β2ARs have been shown to couple to Gi both in vitro (38), and in the heart (36,39,40,41).
Table 11-2 Potential Mechanisms for Decreased β-adrenergic Responsiveness in Failing Human Myocardium | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Given the biological relevance of these receptors, it is not surprising that they have evolutionarily developed a highly regulated mechanism to turn off the signal (42). Rapid waning of receptor responsiveness to agonist stimulation (within seconds or minutes) is dependent on receptor phosphorylation, and results in uncoupling of the receptor from its signal-transducing G-protein. A slow mechanism for receptor desensitization (hours or days) is receptor downregulation (42). In this case a decrease in receptor synthesis, a destabilization of receptor messenger RNA, or an increase in receptor degradation leads to the net loss of cell surface receptors (43,44).
The processes of desensitization and downregulation are initiated by a phosphorylation signal that determines the uncoupling of the receptor from its signal-transducing G-protein and enhances the affinity of the receptor for cytosolic proteins known as the β-arrestins (42). Once bound to the receptor, β-arrestins not only interdict further G-protein coupling and target the activated receptor for endocytosis (45), but also act as a scaffold for the assembly of complex signaling cascades, like the mitogen-activated protein kinase (MAPK) pathway (46,47,48). Once internalized, receptors are targeted to specialized intracellular compartments, where they can be dephosphorylated and recycled to the plasma membrane (early endosomes) or sent down a degradation pathway (late endosomes) (42,49). Interestingly, accumulating evidence suggests that the process of βAR internalization per se may be pathological because the activation of signals arising from internalizing receptors can directly activate maladaptive signaling pathways (50,51). Therefore, strategies that prevent this redistribution may exert a beneficial effect in the failing cardiomyocytes (49,51).
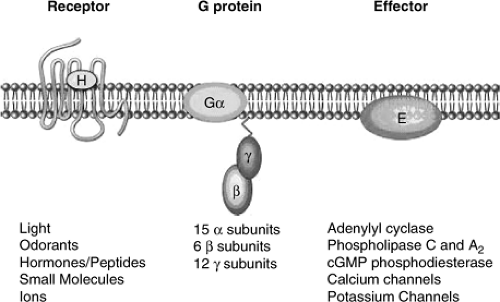 Figure 11-6 Summary of G-protein-coupled receptor signaling. The basic unit of G-protein-coupled receptor signaling comprises a 7-transmembrane-spanning receptor, a heterotrimeric G-protein, and an effector molecule such as an enzyme or ion channel. (Adapted from Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415(6868): 206–212 , with permission.) |
βAR phosphorylation can be mediated by second messenger kinases (for example, protein kinase A or protein kinase C), in a process known as heterologous desensitization or non-agonist-specific desensitization, or by G-protein-coupled receptors kinases (GRKs) (42). The β-adrenergic receptor kinase (βARK1), now commonly known as GRK2, is the most abundant GRK expressed in the heart (52,53). An important binding partner for GRK2 is the multifunctional enzyme phosphoinositide 3-kinase (PI3K). Binding of cytosolic GRK2 to liberated Gβγ subunits of the heterotrimeric G-protein facilitates its translocation to the plasma membrane where it then phosphorylates the agonist-bound receptor (Fig. 11-7) (54). Following agonist stimulation and release of Gβγ subunits, PI3K is recruited to activated βARs by GRK2, where it catalyzes the generation D-3 phosphoinositides that are required for receptor endocytosis (Fig. 11-7) (55,56).
βARs renew their ability to respond to ligands through a process of resensitization, that has been shown to require internalization into intracellular compartments where the acidic environment allows for dephosphorylation of the receptor (54,57). Indeed, it has been recently suggested that a continuous equilibrium between internalization and recycling of βARs to the sarcolemma exists in vivo under physiological conditions, and this plasma membrane-endosome bidirectional trafficking of βARs may be responsible for the resensitization of receptors after prolonged catecholamine exposure (58). Molecular interventions that promote a rapid cycle of receptor dephosphorylation and recycling to the plasma membrane might also represent novel possible therapeutic strategies to normalize βAR function.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


