Fig. 35.1
Baroreflex circuit. The arterial baroreceptors are mechanoreceptors located in the carotid sinuses (innervated by the glossopharyngeal nerve, IX) and aortic arch (innervated by the vagus nerve, X) that respond to stretch elicited by increase in arterial pressure. Primary baroreceptor afferents provide monosynaptic excitatory input to the nucleus of the solitary tract (NTS). Barosensitive NTS neurons initiate a sympathoinhibitory pathway that involves a projection from the NTS to interneurons in the caudal ventrolateral medulla (CVL) that send an inhibitory projection to sympathoexcitatory neurons located in the rostral ventrolateral medulla. The baroreflex-cardioinhibitory pathway involves a direct input from the NTS to a group of vagal preganglionic neurons located in the ventrolateral portion of the nucleus ambiguus (NA). These neurons project to the cardiac ganglion neurons that elicit bradycardia. The baroreflex, via the NTS, also inhibits secretion of arginine vasopressin by magnocellular neurons of the supraoptic (SON) and paraventricular (PVN) nuclei of the hypothalamus, in part by inhibiting noradrenergic cells of the A1 group (Reproduced with permission from Benarroch [7])
More recently, the implication of the SNS in the long-term regulation of BP has also been recognized [8]. A crucial role in this regard seems to be played by the lamina terminalis of the third ventricle of the hypothalamus. This structure is able to sense plasma concentrations of several hormones involved in the regulation of BP and volume, such as angiotensin II (Ang II). The information is integrated and transmitted to the PVH, which sends excitatory stimuli to the sympathetic neurons in the RVLM [7].
35.3 Evidence of Increased SNS Activity in Patients with Essential Hypertension
Early evidence for the role of SNS in the pathogenesis of essential hypertension came from observations in hypertensive individuals exhibiting signs of hyperkinetic circulation and/or high levels of plasma norepinephrine (NE) or urinary adrenergic metabolites [9].
Substantial progress in the understanding of mechanisms of neural hypertension has been made after the introduction of sophisticated techniques for measuring organ-specific sympathetic activity, such as regional NE spillover and microneurography. The regional NE spillover measurement provides information about the activity of sympathetic nerves in organs such as the heart and the kidney. The procedure requires infusion of labeled NE and blood sampling from the coronary sinus (for cardiac NE spillover) or the renal vein (for renal NE spillover) [6]. Microneurography involves the percutaneous insertion of a high-impedance microelectrode, enabling the recording of sympathetic nerve discharges to skin and skeletal muscle vasculature. In humans, the most common measurement is muscle sympathetic neural activity (MSNA), performed at the peroneal nerve [6].
Interestingly, in healthy young persons of both sexes, no relation was found between MSNA and BP. In young men this is explained by the fact that the level of resting MSNA is inversely correlated with cardiac output and vascular reactivity (i.e., higher MSNA is balanced by lower cardiac output and diminished vascular responsiveness to NE). In young women these relationships are absent, but other compensatory mechanisms (including the vasodilator effect of estrogens) may be involved [8]. In contrast, in people older than 40 years, resting levels of MSNA are strongly correlated with resting BP, suggesting that the balance between MSNA and cardiovascular functions is no longer preserved in older persons. In addition, MSNA increases with age and this may further contribute to the increase in the risk of hypertension associated with aging [6].
Studies using both global and regional assessments of SNS activity have shown that sympathetic hyperactivity can be detected in many patients with essential hypertension, including borderline, mild-to-moderate, and severe hypertension, in young, middle-age, and elderly patients, in isolated systolic hypertension and combined systolic-diastolic hypertension, as well as in white-coat and masked hypertension [9]. It is currently estimated that a neurogenic pathogenesis is involved in up to 50 % of all cases of essential hypertension [10]; this estimate is based on both the proportion of untreated patients with essential hypertension who have evidence of sympathetic hyperactivity and the number in whom significant BP lowering is achieved with sympatholytic drugs [11].
Interestingly, it has been observed that the adrenergic overdrive in hypertension parallels the increase in BP, suggesting a cause-effect relationship. In addition, for a similar level of BP, MSNA appears to be more elevated in patients with refractory hypertension than in those who respond well to antihypertensive drug therapy [12].
Important evidence for the involvement of SNS in the pathogenesis of hypertension has been provided by studies with sympatholytic drugs including β-blockers and central α2 receptor agonists (like clonidine and moxonidine), which have long been confirmed as effective antihypertensive therapies (Chap. 39). Furthermore, sympathetic denervation procedures (particularly, renal denervation) have also demonstrated high efficacy in decreasing both sympathetic activity and BP in patients with essential hypertension. Surgical procedures, such as total thoracic sympathectomy, splanchnicectomy, and celiac ganglionectomy, were used several decades ago in the treatment of hypertension, especially in resistant cases, and have resulted in significant lowering of BP, target-organ protection, and increased survival in many patients; however, these surgical methods have been abandoned, because of their invasiveness and high rate of adverse events and mortality [13]. Instead, renal sympathetic denervation has gained increasing popularity in the past 15 years for the treatment of resistant hypertension, due to its proven efficacy and tolerability (Chap. 42). Renal denervation is a percutaneous, minimally invasive procedure, which consists of delivering radiofrequency energy to the renal artery wall from within the vascular lumen. This results in a thermal injury to the renal sympathetic nerves that lie within and next to the renal artery wall [5]. Data collected in different animal models of experimental hypertension (including spontaneously hypertensive rats, stroke-prone rats, Dahl rats, DOCA-salt-induced hypertension, Goldblatt rats, and Ang II- or overfeeding-induced hypertensive rabbits and dogs) have convincingly documented that renal denervation may prevent the development or slow down the progression of hypertension [13]. In hypertensive humans, renal denervation markedly decreases whole-body NE spillover, renal NE spillover, and MSNA [14]. More importantly, clinical trials including the Symplicity Hypertension (HTN)-1 (with extended follow-up) [15] and the Symplicity HTN-2 studies [16] have demonstrated that after renal denervation systolic BP decreases by 20–30 mmHg on average, and this reduction in BP is long lasting, persisting for more than 3 years of follow-up [11]. In addition, other studies have shown that renal denervation significantly decreases left ventricular hypertrophy, improves systolic and diastolic functions, and reduces arterial stiffness, the incidence of proteinuria, and insulin resistance [17]. However, the very recent Symplicity HTN-3 trial, in which 535 patients were randomized single blind to denervation group versus sham-procedure group, did not show a significant reduction of 24-h ambulatory systolic BP [18].
35.4 SNS Hyperactivity and Target-Organ Damage
Sympathetic activation was shown to be more pronounced in hypertensive patients with target-organ damage than in those with uncomplicated hypertension. Thus, sympathetic nerve activity is higher in the presence of renal impairment and appears to increase as renal function worsens. Sympathetic drive is also higher in patients with hypertension complicated with left ventricular hypertrophy, diastolic dysfunction, heart failure, and ventricular arrhythmias [12] (Chap. 5).
The role of the SNS in the development of cardiac hypertrophy has been demonstrated by numerous studies in both animals and humans. In virtually all experimental models, the increase in ventricular wall thickness is associated with increased sympathetic cardiac drive. Chronic systemic infusion of NE induces ventricular hypertrophy, whereas both α-blockade and sympathectomy can reduce the fibrosis associated with myocardial hypertrophy [19].
A few experimental and human studies have shown that vascular remodeling and renal damage in hypertension may also be mediated by the SNS. For example, NE and epinephrine stimulate proliferation of vascular smooth muscle cells (VSMCs) in cultures. In rabbits, carotid artery wall thickness is reduced after local denervation, while in rats, carotid artery distensibility increases after chemical sympathectomy. In humans, sympathetic activity is correlated with BP variability, which is an independent cardiovascular risk factor [12].
35.5 Causes and Mechanisms of SNS Hyperactivity in Hypertension
The specific causes of the increased sympathetic activity in essential hypertension are only partially known. Genetic influences, high salt intake, as well as several metabolic and neurohumoral abnormalities appear to be involved.
35.5.1 Genetic Factors
Spontaneously hypertensive rats exhibit an increased renal SNS activity even before the onset of hypertension [20].
In humans, baroreceptor dysfunction [14] and abnormal increases in plasma NE and whole-body NE spillover have been demonstrated in normotensive individuals with a family history of hypertension [12]. In microneurography studies including normotensive controls, both the number and amplitude of muscular sympathetic bursts were shown to be higher in individuals with a family history of hypertension [14], as well as in those with white-coat [21] and masked hypertension [22]. During mental stress, SNS activity and BP increase in normotensive offspring of patients with essential hypertension, but not in those with normotensive parents [10].
Genetic polymorphisms concerning several genes have been found to be associated with a high prevalence of SNS overactivity and hypertension, including the genes of neuropeptide Y receptor, renalase (amine oxidase that specifically degrades circulating catecholamines), and catestatin (a product of adrenergic neurotransmitter chromogranin A) [23]. Also see Chap. 32.
35.5.2 High Salt Intake
In salt-sensitive hypertensive rats, salt loading increases renal sympathetic activity, which, in turn, may contribute to BP elevation [24]. In deoxycorticosterone acetate (DOCA)-treated rats, salt loading-induced sympathetic stimulation indirectly activates the thiazide-sensitive Na+/Cl− cotransporter (NCC), whereas renal sympathetic denervation leads to the normalization of both NCC activity and BP. The infusion of NE in mice also upregulates NCC, leading to salt-induced hypertension, while treatment with β-blocker propranolol reverses these changes [25]. On a high-salt diet, hypertensive patients with salt-sensitive hypertension have higher plasma NE levels than salt-resistant individuals [25]. Hypertensive humans and animals also develop slightly higher Na+ concentrations in plasma and cerebrospinal fluid after exposure to high-salt diet. It has been suggested that these high Na+ concentrations activate osmoreceptors in the hypothalamic lamina terminalis, which subsequently induces SNS activation. These osmoreceptors are not downregulated by prolonged osmolality changes and thus are able to maintain chronic sympathetic stimulation [10]. Surprisingly, however, in patients with established hypertension, dietary salt restriction can actually lead to an augmentation rather than a decrease of sympathetic activity, as measured by microneurography [12].
35.5.3 Baroreceptor and Chemoreceptor Dysfunctions
In essential hypertension, the function of arterial baroreceptors is altered, exhibiting a decrease in the vagal drive to the heart. In contrast, the sympathetic baroreflex modulation appears to be preserved, although it is reset to a higher level [13]. However, an increased sympathetic drive may result from a reduction in the inhibitory influence of cardiac stretch receptors, in the presence of left ventricular hypertrophy and diastolic dysfunction [12]. Another mechanism that can lead to sympathetic hyperactivation is the impairment of arterial chemoreceptors. Studies based on microneurography have found an exaggerated hypoxia-triggered sympathetic response in patients with hypertension, particularly in those with sleep apnea [12], while deactivation of chemoreceptors with hyperoxia resulted in reductions of MSNA [26].
35.5.4 Obesity and the Metabolic Syndrome
Several studies have shown that chronic SNS activation is particularly increased in patients with obesity- and metabolic syndrome-related hypertension [11]. Central (visceral) obesity is associated with a higher sympathetic activity than subcutaneous obesity [27]. Although there has been a debate as to whether sympathetic activation is a cause or a consequence of obesity, most studies support the view that obesity leads to sympathetic activation [17], which is presumably a compensatory mechanism, aiming to increase energy expenditure [28].
In rabbits, body weight, BP, heart rate, and renal sympathetic nerve activity (RSNA) increased after 1 week of high-fat diet feeding [29]. Similarly, high-fat diets in rats produced an increase in body fat mass, associated with an increase in lumbar sympathetic nerve activity, which became significant by day 12 [30]. Obese dogs receiving a high-fat diet developed sodium retention and hypertension, whereas renal denervation prevented both, indicating that renal nerves have a critical role in obesity-related hypertension [31].
In human obesity, sympathetic overdrive is detectable before hypertension occurs. High levels of circulating catecholamines, MSNA, and kidney NE spillover have been observed in normotensive obese individuals [27]. In one study, MSNA was found to be approximately 40 % higher in obese than in lean normotensive subjects [31]. When obesity and hypertension are both present in the same patient, the degree of sympathetic activation is much greater than in those with either condition separately [27]. On the other hand, weight loss during hypocaloric diet significantly reduces MSNA and whole-body NE spillover rate in obese subjects [32]. In patients with metabolic syndrome, dietary weight loss decreases sympathetic nerve firing and improves hemodynamic and metabolic parameters [33].
However, the activation of the SNS in obesity is not homogeneous. In fact, SNS outflow to the skeletal muscle vasculature and the kidneys is elevated, whereas activation to the heart is reduced, and activation to the skin and splanchnic regions remains unchanged [34]. It is thought that renal sympathetic nerves mediate most, if not all, of the chronic effects of SNS activation on BP in obesity [27]. Several potential factors have been proposed to contribute to increased SNS activity in obesity, including hyperinsulinemia/insulin resistance, hyperleptinemia, hypoadiponectinemia, hypoghrelinemia, increased Ang II levels, obstructive sleep apnea, oxidative stress, inflammation, and baroreceptor dysfunction.
35.5.4.1 Insulin Resistance and Hyperinsulinemia
Insulin resistance is due to the failure of glucose transporter-4 (GLUT-4) to uptake blood glucose into peripheral tissues, especially skeletal muscle; consequently, blood glucose levels remain elevated and continue to trigger insulin release, leading to hyperinsulinemia [34].
In animal studies, intracerebral administration of insulin increases sympathetic outflow, by stimulating the arcuate nucleus, via the PVN [35]. Hyperinsulinemia induces sympathetic activation in normal individuals [31]. Insulin secretion following a meal or during a hyperinsulinemic–euglycemic clamp determines an increase in MSNA and enhances the arterial baroreflex gain of sympathetic activity [27].
However, other studies did not confirm the influence of hyperinsulinemia on sympathetic overactivity and hypertension. For example, the BP does not rise in dogs or in humans treated with insulin [36]. Although acute CNS injection of insulin in rodents may raise RSNA, chronic CNS infusion of insulin does not alter the BP or heart rate [37]. Patients with insulin-resistant metabolic syndrome show blunted sympathetic responses to increased plasma insulin following a glucose load [38]. Finally, patients with insulinoma, in whom fasting insulin levels are four- to fivefold higher than normal, do not exhibit increased SNS activity or elevated BP [39].
35.5.4.2 Hyperleptinemia
Leptin is a protein secreted by adipocytes, in direct relation with fat mass. Leptin acts on multiple CNS centers, especially in the hypothalamus, to inhibit appetite and increase thermogenesis. Thus, leptin’s function is to maintain adipose tissue mass stability through a negative feedback mechanism [40].
Leptin also contributes to the regulation of BP, through its ability to stimulate SNS centers in the hypothalamic arcuate nucleus and the NTS [27]. Transgenic mice that overexpress leptin have elevated BP compared with normal controls [41]. In rats, leptin administration was shown to increase sympathetic outflow to the kidneys, adrenal glands, and limbs. Chronic increases in systemic or CNS leptin concentrations induce BP elevation in rodents, which can be prevented by adrenergic receptor blockade [31]. Diet-induced obesity in mice is associated with hyperleptinemia and elevated SNS activity and BP [42]. In humans, plasma leptin concentration positively correlates with RSNA, as well as with the development of hypertension [43]. Several epidemiological studies have shown positive correlations between circulating leptin and BP levels, independent of obesity [31].
In obese animals and patients, although resistance to the anorexigenic action of leptin has been clearly documented, leptin’s sympathetic effects are preserved [44]. Thus, despite failing to regulate food intake and body weight, leptin appears to have a significant role in the development of hypertension in obesity [42].
35.5.4.3 Hypoadiponectinemia
Adiponectin is a cytokine synthesized by adipose tissue, which regulates lipid and glucose metabolism. High levels of adiponectin improve insulin resistance, dyslipidemia, and atherosclerosis [31].
Adiponectin also reduces efferent sympathetic nerve traffic to the kidney and decreases BP in rodents. Higher circulating adiponectin levels are associated with lower BP in humans. On the other hand, metabolic syndrome and hypertension are associated with low adiponectin levels. Furthermore, subjects with hypoadiponectinemia exhibit cardiac sympathetic overactivity. Taken together, these findings suggest that low adiponectin may play a role in the pathogenesis of obesity-associated hypertension [31].
35.5.4.4 Hypoghrelinemia
Ghrelin is a peptide produced by the stomach, which rises during fasting and appears to play a key role in triggering hunger. In addition, acute ghrelin infusion into the CNS of rodents reduces SNS activity and counteracts the stimulatory effects of leptin. Ghrelin levels are reduced in obesity, perhaps as a compensation for excess energy intake [40]. Thus, it seems plausible that the low ghrelin concentrations observed in obesity could contribute to hypertension.
35.5.4.5 Angiotensin II
Increased Ang II levels in obesity have also been suggested to cause SNS activation. In two studies in rabbits, Ang II treatment produced a modest rise in BP but a marked increase in RSNA [45]. Ang II-induced oxidative stress in the RVLM was shown to induce sympathoexcitation in rats with obesity-related hypertension [28]. Hilzendeger et al. showed that the intracerebroventricular administration of losartan, an Ang II receptor blocker, inhibited leptin-mediated increases in RSNA, suggesting that Ang II may also facilitate sympathetic activation by leptin [46]. Acute Ang II infusion in normotensive humans raises MSNA, while renin–angiotensin system inhibition reduces it [28]. In a study by Grassi et al., the blockade of Ang II for 3 months in patients with metabolic syndrome decreased MSNA by 21 % [47].
35.5.4.6 Obstructive Sleep Apnea
In patients with obstructive sleep apnea (OSA), MSNA is increased, even during the awake state. Patients with OSA are often obese, and MSNA is higher in obese persons with OSA than in those without OSA. Sympathetic activation associated with OSA is due to chemoreflex mechanisms stimulated by hypoxia and hypercapnia, as well as to altered baroreflex responsiveness. This sympathetic activation, along with other mechanisms, contributes to the increased risk of hypertension and cardiovascular disease seen in these patients [6]. OSA can be complicated by pulmonary hypertension, which has also been associated with increased MSNA [48]. Nevertheless, sympathetic hyperactivation in patients with OSA can be partially or completely reversed by long-term continuous positive airway pressure therapy [6].
35.6 The Renin–Angiotensin System in BP Regulation
The renin–angiotensin system (RAS) is a key regulator of BP and fluid homeostasis. Hyperactivation of the RAS plays a central role in the pathogenesis of several forms of secondary hypertension, such as renin-secreting tumors, renal artery stenosis, and renal microvascular diseases. In addition, there is evidence that RAS is involved in essential hypertension, as well as in vascular, cardiac, and renal damage associated with hypertension and other disorders [49].
The RAS cascade begins with the synthesis of renin by the juxtaglomerular (JG) cells in the kidney. The release of renin into the circulation depends on four mechanisms: (1) a renal baroreceptor mechanism in the afferent arteriole that senses changes in renal perfusion pressure, (2) changes in the delivery of NaCl to the macula densa cells of the distal tubule (which lie close to the JG cells and, together, form the “JG apparatus”), (3) sympathetic nerve stimulation via β-1 adrenergic receptors, and (4) negative feedback by a direct action of angiotensin II (Ang II) on the JG cells. Renin secretion is stimulated by a decrease in perfusion pressure or in NaCl distal tubule delivery and by an increase in sympathetic activity [49].
Renin controls the initial, rate-limiting step of the RAS by cleaving the N-terminal portion of angiotensinogen, a high-molecular-weight globulin, mainly secreted by the liver, to form the biologically inactive decapeptide Ang I or Ang-(1–10). Ang I is further hydrolyzed by the angiotensin-converting enzyme (ACE), which removes the C-terminal dipeptide to form the octapeptide Ang II [Ang-(1–8)], the active effector of the RAS (Chap. 36). ACE is a membrane-bound carboxypeptidase, located on various cell types, including vascular endothelial cells (ECs), brush border epithelial cells, and neuroepithelial cells [49].
Ang II acts on many tissues and organs, including blood vessels, kidney, heart, and brain, through binding and activation of receptors, which belong to the large family of G protein-coupled (GPCR) or 7 transmembrane (7TM) spanning receptors. At least four angiotensin receptor subtypes have been described (AT1R to AT4R) [49].
A summary of the RAS, including the Ang peptide family and physiological effects mediated via ATR subtypes, is presented in Fig. 35.2 [50].
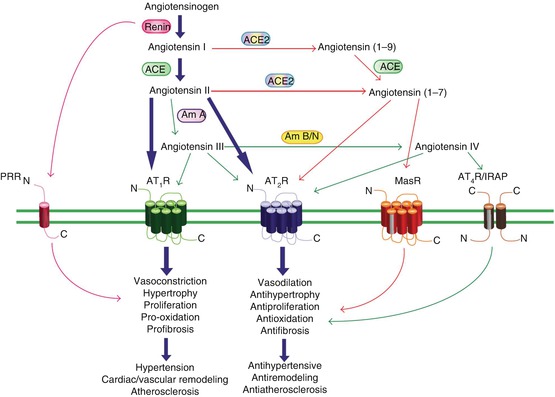
Fig. 35.2
Summary of the RAS incorporating the Ang peptide family and physiological effects mediated via ATR subtypes. Under the classical RAS schema, Ang II is produced, via renin and ACE, to act with equal affinity on two ATR subtypes, AT1R and AT2R (large arrows). However, it is now appreciated that a number of breakdown products of Ang II, namely, Ang (1–7), Ang III, and Ang IV, exert their own unique effects that are distinct (and often opposite) to those of Ang II. Such effects are often mediated via newly recognized receptors such as MasR for Ang (1–7) and AT4R (also known as IRAP) for Ang IV or additionally via AT2R stimulation. ACE2 is also a new pathway for the formation of Ang (1–7). Newly identified Ang receptor-binding proteins associated with different ATR subtypes may also modify ATR activation. Thus, overstimulation of AT1R (and PRR) by Ang II, which can contribute to a plethora of cardiovascular disease processes, may be counter-regulated by a number of non-AT1R mechanisms. Most notably, AT2R stimulation usually causes opposing effects to AT1R, as indicated. It is also likely that the MasR exerts a similar counter-regulatory role, whereas the evidence is more preliminary and speculative for AT4R/IRAP. In terms of mediators, Ang II itself stimulates AT2R, whereas the shorter Ang peptides stimulate their cognate receptors and possibly also AT2R. (Am A aminopeptidase A, Am B/N aminopeptidase B/N, IRAP insulin-regulated aminopeptidase, PRR prorenin receptor) (Reproduced from Elton et al. [50] (an open access article))
35.6.1 AT1R-Mediated Effects of Ang II
The main physiological function of Ang II is to maintain BP and volume homeostasis. This is achieved by several mechanisms, including vasoconstriction of renal and systemic arterioles, stimulation of renal tubular sodium reabsorption, and release of aldosterone from the adrenal glands. All these effects are mediated by AT1 receptors (AT1R) [51]. In states of hypotension and volume depletion, Ang II acts as a rapid and potent vasoconstrictor, by activation of phospholipase C (PLC) pathway in vascular smooth muscle cells (VSMCs) [52].
In the kidney, the activation of apical Na+/H+ exchange, basolateral Na+/HCO3 − cotransport, and basolateral Na+/K+-ATPase and apical H+-ATPase are implicated in Ang II-induced sodium and bicarbonate reabsorption in the proximal tubule, whereas Na+/H+ exchange and H+-ATPase contribute to reabsorption of sodium and bicarbonate in distal tubules [53]. Hypertensive patients display an enhanced antinatriuretic response to exogenously administered Ang II [54].
Ang II has been shown to induce NE discharge at the neurovascular junction, in the kidneys, adrenal glands, and CNS [55]. In the brain, Ang II activates the SNS by promoting oxidative stress and the release of an endogenous digitalis-like factor (EDLF) [56], whereas RAS inhibitors can suppress sympathetic hyperactivity [55]. Ang II also stimulates thirst, salt appetite, and vasopressin secretion, by binding to AT1R in the subfornical organ, the median preoptic nucleus, the PVN, and the supraoptic nucleus [57].
Moreover, Ang II is presently recognized not merely as a BP regulator but as a pleiotropic hormone, which contributes to the development and progression of vascular, cardiac, and renal damage associated with hypertension and other diseases. These deleterious effects are mainly mediated by AT1R, via several intracellular signaling systems, including G protein-dependent [like protein kinase C (PKC), ERK1/2, Raf, tyrosine kinases, nuclear factor κB (NFκB), and reactive oxygen species (ROS)] and independent (such as β-arrestin-mediated MAPK activation and Src-JAK/STAT) pathways. The AT1R-mediated activation of NADPH oxidases (NOX) releases ROS, resulting in the activation of proinflammatory transcription factors and stimulation of small G proteins such as Ras, Rac, and RhoA [58].
Proinflammatory, profibrotic, and prooxidant effects of Ang II on the cardiovascular system have been demonstrated in numerous animal and human studies [59]. For example, it has been shown that Ang II induces increased expression of inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF- α) in the heart [60], arteries [61, 62], and peripheral monocytes [63]. Ang II also upregulates chemokines, like monocyte chemoattractant protein-1 (MCP-1) [61, 62], and adhesion molecules, including vascular cell adhesion molecule 1 (VCAM-1) [62] and intercellular adhesion molecule-1 (ICAM-1) [64], thereby inducing activation and infiltration of monocytes and other leukocytes into the heart and vessel wall [61, 64]. Furthermore, Ang II contributes to oxidative stress, by stimulating NOX, which produce superoxide and hydrogen peroxide, and by increasing the production of mitochondrial ROS [65]. Ang II also facilitates vascular calcification, by reducing the expression of matrix Gla protein (MGP), an inhibitor of calcification, in VSMC [66]. All of these effects are mediated by AT1R and can be prevented by administration of AT1R antagonists [61, 62, 64].
In animal models of hypertension, elevated cardiac Ang II enhances cardiac myocyte apoptosis, macrophage infiltration, and expression of NOX2 and transforming growth factor-β1 (TGF-β1), thus contributing to left ventricular hypertrophy and fibrosis [67]. The inflammatory and profibrotic effects of Ang II seem to be regulated by cytosolic adaptor caspase recruitment domain 9 (CARD9) [68] and by C-reactive protein [69]. Interestingly, studies of Crowley et al. in a mouse kidney cross-transplantation model have shown that Ang II infusion is capable to induce cardiac hypertrophy only in the presence of renal AT1R; when AT1R are deleted from the kidney, the residual repertoire of systemic, extrarenal AT1R is not sufficient to induce hypertension or cardiac hypertrophy. This suggests that Ang II causes cardiac hypertrophy not by direct effects on cardiac AT1R but by increasing the BP through activation of renal AT1R [70].
In the kidney, besides vasoconstriction and sodium reabsorption, Ang II induces inflammation, renal cell growth, mitogenesis, apoptosis, migration, and differentiation [71]. Infusion of Ang II in mice results in increased NOX activity, increased expression of proinflammatory cytokines and fibrosis-associated genes (such as α-smooth muscle actin, TGF-β, procollagen type I-α1), and increased levels of collagen I, with histological evidence of tubulointerstitial fibrosis [72]. In animal models of glomerular disease, Ang II induces oxidative stress, expression of adhesion molecules, macrophage infiltration, and exacerbation of proteinuria [73]. These effects were also shown to be mediated by AT1R and prevented by AT1R blockade.
In the cerebral circulation, Ang II has been involved in ROS generation, alterations of vasomotor function, impaired neurovascular coupling, inflammation, and vascular remodeling [74]. Ang II infusion in rodents increases ROS production by cerebral arteries [75], via activation of AT1R and NOX2 [76]. Ang II also induces constriction of cerebral arteries, mediated by AT1R on VSMC and EC [77], and impairs endothelium-dependent vasodilation by reducing the bioavailability of NO and potentially by interfering with other endothelium-dependent mechanisms [77]. Ang II-induced hypertension is associated with impairment of neurovascular coupling (i.e., regulation of local cerebral blood flow in response to changes in metabolic demands that result from increased brain activity) [75]. Furthermore, Ang II causes remodeling and hypertrophy of cerebral arterioles, possibly mediated by NOX2 [78]. In spontaneously hypertensive rats, AT1R blockade reduces the expression of ICAM1 and macrophage infiltration in the brain, suggesting a key role for Ang II in cerebral inflammation [79]. Increased leukocyte and platelet adhesion has been observed during Ang II-induced hypertension, dependent on AT1R on these cells [80]. On the other hand, several studies in hypertensive patients have demonstrated that treatment with ACE inhibitors and, especially, with AT1R blockers is capable to improve total cerebral blood flow [74].
35.6.2 The Role of Aldosterone in Hypertension
The implication of elevated aldosterone levels in hypertension has been recognized long ago, in the setting of primary hyperaldosteronism, initially described by Conn in 1955 [81].
Aldosterone is secreted by the zona glomerulosa of the adrenal cortex under the influence of Ang II (via AT1R), serum potassium, and adrenocorticotropic hormone (ACTH). In the kidney, aldosterone binds to the mineralocorticoid receptors (MRs) in the epithelial cells of the distal convoluted tubules and collecting ducts. By upregulating the basolateral Na+/K+ exchange pump, the epithelial sodium channels (ENaCs), and the outer medullary renal K channels, aldosterone promotes sodium and water reabsorption and secretion of K into the tubular lumen. These effects are mediated by both genomic and nongenomic (rapid) pathways [81]. In addition, it has been recently shown that aldosterone can directly induce vasoconstriction, via MR on VSMC [82]. Aldosterone can also activate the SNS by central mechanisms [83], thus further contributing to the increase of BP.
Besides its antinatriuretic and vasopressor effects, aldosterone is involved in vascular and target-organ damage, by promoting oxidative stress, inflammation, and fibrosis of the heart, vasculature, and kidneys, through both MR-dependent and MR-independent pathways [81]. Aldosterone induces the formation of ROS through several mechanisms: (1) stimulation of NOX activity in macrophages, VSMC, EC, cardiomyocytes, podocytes, and mesangial cells, (2) decrease of glucose-6-phosphate dehydrogenase in vascular cells, and (3) stimulation of mitochondrial production of ROS. Oxidative stress in the vasculature induces endothelial dysfunction and VSMC proliferation and migration [82].
Direct activation of MR in VSMC and EC has been shown to promote inflammatory gene expression [82]. In mineralocorticoid-induced hypertension models, MR activation has been associated with perivascular inflammatory cell infiltration; increased expression of proinflammatory factors including ICAM1, MCP-1, and cytokines in cardiac tissue [84]; and increased expression of osteopontin, MCP-1, IL-6, and IL-1β in the kidney [85]. MR activation in human EC promotes expression of adhesion molecules ICAM1 and VCAM1, resulting in enhanced leukocyte adhesion to human coronary EC [86]. MR antagonism reduces the vascular inflammation and ameliorates cardiac and renal injury, even without changes in BP, supporting that MR activation participates in vascular inflammation and damage through a BP-independent, direct vascular process [87]. In clinical studies, infusion of aldosterone or Ang II into healthy subjects increases circulating IL-6 concentrations, and the Ang II effect is blocked by spironolactone, suggesting an MR-dependent mechanism [88]. Treatment with spironolactone has also been shown to reduce MCP-1 and plasminogen activator inhibitor (PAI-1) levels in subjects with type 2 diabetes and hypertension, respectively [89].
Aldosterone also stimulates the expression of profibrotic molecules, such as transforming growth factor-β1 (TGF-β1), PAI-1, endothelin 1 (ET-1), placental growth factor (PGF), connective tissue growth factor (CTGF), osteopontin, and galectin-3 [89]. In cultured VSMC, aldosterone stimulates collagen and fibronectin synthesis [90]. In vivo, chronic administration of aldosterone causes interstitial and perivascular fibrosis in the heart, fibrosis and stiffness of the aorta, and glomerulosclerosis and interstitial fibrosis in the kidney [89]. Patients with primary aldosteronism were found to have significantly increased vascular medial thickness and narrowed vessel lumens, compared to patients with similar degrees of essential hypertension and other forms of secondary hypertension [82]. In the Framingham Offspring Study, the aldosterone/renin ratio was positively correlated with left ventricular hypertrophy and with the development of arterial stiffness [91].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


