Surgical Myocardial Protection
Jakob Vinten-Johansen
Neil J. Thomas
INTRODUCTION
The science of myocardial protection had its beginnings in the 1950s, and has enjoyed a robust and often controversial development since. Great strides were made in the 1980s and 1990s which shaped the cornerstones of the cardioplegia strategy that we largely use today (crystalloid or blood, warm, tepid or cold, continuous or intermittent, antegrade or retrograde). Mortality has substantially decreased over the past decades as a result of applying cornerstone as well as novel cardioprotective strategies and new technologies.
However, the patient of the 1980s and 1990s during which cardioplegia was developed has morphed considerably in the 2000s. Active lifestyles have changed to more sedentary lifestyles beginning in the early school years with less emphasis placed on physical activity, and more distractions from active lifestyles imposed by the digital-age gadgets. This, coupled with the availability of fast food with high caloric, high fat, and high sodium content, has conspired to radically change our body habitus and biology; indeed, diabetes, obesity, hypercholesterolemia, and the metabolic syndrome are more prevalent today. These more prevalent comorbidities have impacted the efficacy of modern surgical therapies and the outcomes (1) of myocardial protection strategies. Experimental animal models in which effective cardioplegia strategies were developed decades ago, and observed to be effective in the healthier patients with normal myocardial tissue substrate preoperatively in which they were initially tested clinically seems to apply in fewer and fewer patients today. The “one formula-fits-all” strategies of the past must be rethought in the current era. With patients living longer as well, advancing age and the more senescent myocardium can display altered responsiveness to some therapeutics. So we may ask, is myocardial protection developed in the 1980s and 1990s being used as effectively on the patient of the 2010s? And, furthermore, the yardstick by which our results are measured is changing in the world of health care today, where a 30-day surgical mortality rate may no longer be the gold standard. Our results must now be viewed in terms of longer-term, event-free survival and with measure of long-term myocardial performance after an index event or surgery.
The concepts of cardioprotective strategies have changed significantly as our understanding of the pathophysiologic determinants of ischemia, and particularly the complex mechanisms involved in reperfusion, have expanded. Early empiric, largely physiologic observations have given way to complex biochemical and molecular interactions, which in some cases have reinforced physiologic intuition and in other cases have redirected our thinking, but has nonetheless provided a scientific underpinning to a more molecular approach in developing target-specific strategies of myocardial protection. This chapter will discuss the pathobiology of ischemia-reperfusion injury, provide the basis by which the severity and duration of evolving ischemic conditions set the stage for reperfusion and oxygen-related injury and its impact on affected tissues as a basis for understanding cardioprotective strategies to attenuate injury. It will also discuss new, emerging concepts of myocardial protection that have developed out of a better understanding of the pathobiology of ischemia-reperfusion injury, and a growing appreciation that the preoperative condition of the tissue may be equally, if not more important, than any intraoperative strategy employed in previously normal tissue. It will also discuss the all-elusive “golden ring” of arresting the heart in a nondepolarized state.
The goal of this chapter is to provide the reader with scientific and practical knowledge of myocardial protection that can be applied to design appropriate strategies to meet the need of specific patients, their unique demographics and clinical scenarios, and to provide flexibility in the approach based on both preoperative condition and intraoperative events. It will also provide some guided reading to enhance the background of relevant topics
HISTORICAL PERSPECTIVES ON MYOCARDIAL PROTECTION AND REPERFUSION INJURY
The need to protect the myocardium from intraoperative and postoperative damage was realized before the development of cardiopulmonary bypass (CPB). Temporary inflow occlusion techniques to limit blood flow to the heart permitted the
repair of simple intracardiac defects, and in many cases the occlusion time could be increased by adding moderate systemic hypothermia (30°C) (2). After the development of CPB by Gibbon and colleagues in 1954 (3,4), profound systemic hypothermia could be used safely to provide a hemodynamic safety net, and to provide protection of the heart and other body organs by permitting longer periods of ischemic cardiac arrest for the repair of more complicated lesions.
repair of simple intracardiac defects, and in many cases the occlusion time could be increased by adding moderate systemic hypothermia (30°C) (2). After the development of CPB by Gibbon and colleagues in 1954 (3,4), profound systemic hypothermia could be used safely to provide a hemodynamic safety net, and to provide protection of the heart and other body organs by permitting longer periods of ischemic cardiac arrest for the repair of more complicated lesions.
The development of extracorporeal circulation techniques allowed cardiac procedures to proceed without threat of hemodynamic collapse. However, performing complicated procedures on the heart was made difficult by its continuous motion and blood in the field. Ischemia was known to achieve cardiac arrest (ischemic or anoxic arrest) through the depletion of high-energy phosphate stores on which cardiac contraction depends. However, myocardial ischemia led to necrosis and contractile dysfunction upon reperfusion, the worst of which was an irreversible “stone” heart (5,6). The conundrum of surgeon convenience at the expense of acceptable outcomes provided a driving force for innovation.
Once the severe complications of ischemic arrest were recognized, chemical methods were used to arrest the heart. Based on reports from Ringer in 1883 that high concentrations of potassium would arrest the heart (7), Melrose and associates in 1955 (8) and 1957 (9) used hyperkalemia to induce asystole during CPB. The combination of immediate arrest and hypothermia would ostensibly reduce energy (i.e., oxygen) demands of the heart, and thereby avoid injury imposed by hypothermia alone. Melrose made a hyperkalemic solution by mixing potassium citrate solution (77 mM) with 18 mL of blood, which was then infused by hand-held syringe into the aortic root proximal to the aortic cross-clamp. This hyperkalemic arresting solution was later called “cardioplegia” solution by Lam et al. (10) and Sergeant et al. (11). Other contemporaries of Melrose used alternative arresting agents, such as acetylcholine alone (10,11) or in combination with other drugs (12).
Although asystole was indeed achieved with the Melrose potassium citrate solution, the concept of elective cardiac arrest was abandoned in the United States for some 20 years following reports of high morbidity and pathologic complications in chemically arrested hearts (13). Chemical cardioplegia was supplanted by alternative techniques of arresting the heart or limiting blood flow in the surgical field, including ventricular fibrillation, intermittent aortic cross-clamping, and profound systemic (14) or topical hypothermia (15). Laboratory studies demonstrated that topical hypothermia protected inadequately against intraoperative injury, which resulted in postoperative myocardial subendocardial necrosis, and postischemic metabolic and functional depression (16,17,18). In the late 1960s, reports described scattered myocardial or subendocardial necrosis in patients who had died after otherwise technically successful cardiac surgical operations, suggesting that the current techniques of myocardial protection were inadequate (19,20). This set the stage for a later re-emergence of chemical cardioplegia in the United States.
During the surgeon-imposed moratorium on the use of the Melrose solution, cardioplegia solutions continued development and clinical use in Europe. Bretschneider et al. (16) and Kirsch et al. (17) used crystalloid formulations which mimicked intracellular ionic concentrations (intracellular crystalloid solutions). The Bretschneider HTK (histidine, tryptophan, α-ketoglutarate)-ketoglutarate) solution used low calcium, low sodium, and procaine with histidine buffers to achieve a nondepolarized arrest, rather than hyperkalemia-induced depolarized arrest. In the 1970s, Hearse and associates (18,21) at St. Thomas’ Hospital in London developed an extracellular hyperkalemic solution based on a rigorous series of experiments that established a scientific understanding of the determinants of injury incurred during global ischemia. Braimbridge introduced St. Thomas’ cardioplegia solution into clinical use in 1975 (22). St. Thomas’ solution was later modified based on further experimental studies to the St. Thomas’ solution No. 2 (23), an intracellular ionic concentration, normocalcemic, and hyperkalemic (16 mmol/L) solution; it is currently marketed as Plegisol in the United States. The Bretschneider HTK solution is currently marketed as Custodiol HTK solution.
Chemical cardioplegia was revived in the United States by Gay and Ebert (24) and Tyers et al. (25,26), who found that the constituents in the Melrose formulation were inappropriate rather than the concept of hyperkalemic chemical cardioplegia itself being erroneous. These reports popularized the use of potassium-based cardioplegia to achieve electromechanical arrest. In the late 1970s and early 1980s, Follette and colleagues from Dr. Buckberg’s laboratory (see reference 224 below) introduced the concept of cold hyperkalemic blood cardioplegia. The physiologic attributes of blood, including its superior buffering capacity, endogenous oxygen radical scavengers, detoxifying substances, and superior oxygen-transport capacity, has established this concept as a popular strategy that has facilitated surgery and vastly improved patient outcomes. Today, a variety of both crystalloid and blood cardioplegia solutions are used clinically worldwide to achieve elective cardiac arrest and a bloodless field.
The cornerstones of myocardial protection that had been laid by the end of the early 1980s are: (1) rapid chemical arrest to conserve high-energy phosphate stores and reduce ischemia, (2) hypothermia to reduce the metabolic rate, (3) adjunctive agents to reduce the effects of ongoing ischemia (anti-ischemic agents), and (4) continuous or intermittent delivery to restore or maintain tissue oxygenation. Further developments in cardioplegia-related myocardial protection strategies focused on (1) optimizing delivery (retrograde, gentle cardioplegia delivery pressures), (2) overcoming the adverse effects of hypothermia, (3) identifying metabolic enhancements (ATP generation and pH management) and buffers, (4) managing calcium (Ca2+) accumulation, and (5) understanding the pathophysiology
of ischemia and reperfusion, which would drive further developments of strategies to avoid these mechanisms of both ischemic and postischemic patterns of injury. There has also been a persistent search for agents that induce polarized arrest which overcome the disadvantages of hyperkalemia (reviewed in Dobson et al. (27) and Maruyama et al. (28)). With the recent rapid developments in molecular and cellular biology, experimental focus has shifted away from the organ and physiologic aspects toward cellular and molecular areas to improve the understanding of the effects of ischemia-reperfusion injury, inflammation, and cell death processes. Understanding the mechanisms involved in the transition to cell death would direct developments in Ca2+ handling and preventing Ca2+ dyshomeostasis, the role of oxidant injury, and a newly recognized role of mitochondria in determining the transition from reversible to irreversible injury, and whether the cell pursues a necrotic pathway or apoptotic pathway to cell death. Finally, the concept of conditioning, which recruits complex endogenous biologic mechanisms of self-protection against stressors such as ischemia-reperfusion injury, oxidants and apoptosis, has been incorporated into pretreatment (preconditioning), intraoperative (preconditioning), and reperfusion (postconditioning) phases of cardiac surgery. Although human beings have been evolving for hundreds of thousands of years, it has only been in the past 60 years or less that the human heart has been subjected to the kind of induced ischemia and surgical reperfusion discussed in these pages.
of ischemia and reperfusion, which would drive further developments of strategies to avoid these mechanisms of both ischemic and postischemic patterns of injury. There has also been a persistent search for agents that induce polarized arrest which overcome the disadvantages of hyperkalemia (reviewed in Dobson et al. (27) and Maruyama et al. (28)). With the recent rapid developments in molecular and cellular biology, experimental focus has shifted away from the organ and physiologic aspects toward cellular and molecular areas to improve the understanding of the effects of ischemia-reperfusion injury, inflammation, and cell death processes. Understanding the mechanisms involved in the transition to cell death would direct developments in Ca2+ handling and preventing Ca2+ dyshomeostasis, the role of oxidant injury, and a newly recognized role of mitochondria in determining the transition from reversible to irreversible injury, and whether the cell pursues a necrotic pathway or apoptotic pathway to cell death. Finally, the concept of conditioning, which recruits complex endogenous biologic mechanisms of self-protection against stressors such as ischemia-reperfusion injury, oxidants and apoptosis, has been incorporated into pretreatment (preconditioning), intraoperative (preconditioning), and reperfusion (postconditioning) phases of cardiac surgery. Although human beings have been evolving for hundreds of thousands of years, it has only been in the past 60 years or less that the human heart has been subjected to the kind of induced ischemia and surgical reperfusion discussed in these pages.
PATHOPHYSIOLOGY OF SURGICAL ISCHEMIA-REPERFUSION INJURY
Because many factors affect postoperative outcomes of hearts with various and complex preoperative pathophysiologic features, developing strategies that adequately protect all hearts during cardiac surgery is often problematic. The heart under consideration may present a complex picture of preoperative disease, variable age, gender, metabolic and cellular dysfunction, global or regional ischemia (both acute and chronic), each of which contribute to differing vulnerabilities to ischemia and reperfusion injuries. However, adequate myocardial protection must be based on a sound scientific basis. Obviously, a single cardioprotective strategy may be inadequate to suit all surgeon preferences and target all patient pathologies. Hence, the surgical team must often mold the composition of the cardioplegic solution or its modality of delivery to meet the requirements of the patient’s pathologic profile as presented. The measures taken to protect the heart during elective cardiac arrest should represent a balance struck between the requirements of the heart during aortic cross-clamp or reperfusion, with appropriate adjustments made for special considerations (hypertrophy, diffuse coronary disease), and avoid distraction from the surgical procedure. Ischemia and reperfusion injury (29) and the choice of cardioplegia technique (30) represent primary contributors to patient outcomes.
Ischemic Injury
When Does Ischemia and Reperfusion Injury Occur during Cardiac Surgery?
It is critical for the surgical team to understand that the presence of acute, ongoing and evolving or progressing ischemia must impact the protection strategy. Ischemia-reperfusion injury can occur in the surgical setting at three major time points during the surgical procedure (summarized in Fig. 9.1). (a) Before CPB has been instituted or cardioplegia solution has been delivered: “Unprotected” clinical ischemia before CPB usually results from the presenting acute coronary syndrome and can be exacerbated by hypotension, arrhythmias (e.g., atrial or ventricular fibrillation), and other hemodynamic changes (e.g., cardiogenic shock, coronary artery spasm). In most cases, acute ischemia is active in both the distribution of the principally involved artery (cardiologists call this the “infarct artery”) but also distributions in which preexistent collateral flow may be compromised. Patients referred for bypass surgery in these circumstances almost always have multi-vessel disease patterns making the myocardial substrate dangerous from the standpoint of the severity and duration of the syndrome, which can be extremely difficult if not impossible to determine. Reperfusion and reoxygenation injury occur when these conditions change abruptly before effective measures are implemented. Recent attention has been paid to avoiding over-oxygenating such patients systemically, and this will be discussed in more detail in later sections.
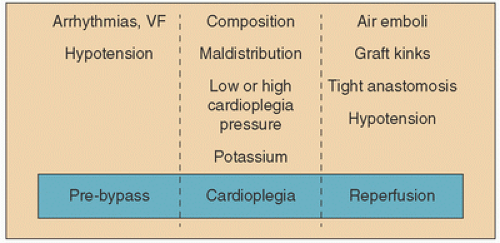 FIGURE 9.1. Events occurring before the institution of cardiopulmonary bypass, during delivery of cardioplegia and at reperfusion that predispose the myocardium to ischemia and reperfusion injuries. Arrhythmias, ventricular fibrillation (VF) or severe hypotension can cause antecedent global ischemia. During the delivery of cardioplegia, the composition of the solution can cause edema, further ischemia if oxygen and nutrients are inadequate to meet ongoing albeit lower demands, and Ca2+ concentration is either too high or too low. Coronary obstructions, air or low cardioplegia infusion pressure can lead to maldistribution of solution. Too high infusion pressure can lead to microvascular injury and edema. Potassium itself has negative effects secondary to placing the cell in a depolarized state. Ischemia may be encountered if air emboli, graft kinks, tight anastomoses, or systemic hypotension cause inadequate distribution of blood flow. Finally, reperfusion injury can cause reversible (stunning, arrhythmias, edema) or irreversible (necrosis, apoptosis) injury. |
(b) At the initiation of cardiopulmonary bypass: At the moment bypass is initiated, blood is immediately diverted from the right atrial drainage system into a venous reservoir where it is then pumped through the oxygenator. This configuration does more to the heart than merely oxygenate venous blood and send it back to the ascending aorta. As the heart is emptied into the venous side of the pump, the oxygen demand of the metabolizing myocardium decreases by 50%, but the high oxygen content of blood extracorporeally oxygenated makes the tissue vulnerable to reoxygenation damage if no steps are taken to mitigate this injury. This might be viewed as an abrupt interruption of antecedent ischemic conditions. While this is one of the principal goals of bypass (to give the heart a “rest”), it must be recognized that the heart is also vulnerable to a pattern of injury that has been, at best, underappreciated in the past. Ihnken et al. (31) suggested as early as 1998 that initiation of bypass is also, perhaps paradoxically, a dangerous moment for the heart. In this work, he observed clear, reproducible biochemical evidence of injury in samples of blood drawn from the coronary sinus. Since Buckberg (32) published his work on the reoxygenation injury in 1995, it has been observed that abrupt over-exposure of the ischemic heart to oxygen increases injury in a pO2-dependent pattern of injury that can be mitigated by altering the oxygen tension of the perfusate.
(c) During the “protected” period of arrest and delivery of cardioplegia: Ischemia or reperfusion injury can occur during the cardioplegia phase. Ischemia can develop or be worsened under several circumstances (1) the unintentional maldistribution of cardioplegia solution distal to stenotic or totally occluded coronary arteries, (2) between intermittent infusions of cardioplegia solution, (3) during interruption of continuous cardioplegia strategies, (4) inadequate delivery of retrograde cardioplegia to areas of the right heart whose venous blood drains directly into the Thebesian system, or areas that are simply under-filled due to malposition of a retrograde delivery device. Though the cardioplegia phase is considered “protected time,” the potential for worsening of, or the development of, ischemic conditions followed by reperfusion injury can occur with the initial administration of cardioplegia and with each subsequent delivery of cardioplegia solution [(re)perfusion] for a variety of reasons: (1) excessive pressure (microvascular injury), (2) low onconicity favoring extravasation of fluids into the extracellular space, (3) formation of oxygen radicals from over-oxygenation and oxidative stress, and (4) other mechanisms of reperfusion injury as discussed below. While delivery of cardioplegia should provide protection, this phase also offers opportunity for additional injury to occur unintentionally. Any surgeon who has been faced with postoperative low-cardiac output and who suspects inadequate protection knows this to be true.
(d) After release of the cross-clamp and attempts to reanimate the heart: Additional ischemia may be encountered at the release of the cross-clamp when (1) coronary blood flow is impaired through kinked grafts or (2) tight anastomoses (3) air emboli are present in the coronary arteries, (4) ventricular fibrillation occurs at low perfusion pressures, or (5) poor ventricular performance or dysrhythmias cause hypotension once the heart is converted and off bypass. Clearly, when the clamp is removed reperfusion injury can occur as blood flow is restored and oxygen exposure is reestablished after any of the above events. In terms of surgically induced ischemia, this is intuitively the major time when one thinks of reperfusion injury occurring.
Understanding the pathophysiologic mechanisms of myocardial injury occurring at these intervals and applying the principles of myocardial protection will help avoid injury induced by the very techniques intended to preserve the myocardium. Unlike the cardiologist, the surgical team has the opportunity to intervene directly to control many facets of ischemia and reperfusion injury, and to limit at some point the mechanisms causing injury, thereby more favorably directing the outcome of the postischemic heart. The pathophysiology of ischemia and reperfusion is described in the following sections. Interventions targeting these various pathologic processes are discussed under Myocardial Protection Therapy.
Determinants of Ischemic Injury
Ischemia encountered either before arrest or during cardioplegic arrest, is a major cause of postcardioplegia injury. Ischemia sets the stage for reperfusion injury; without ischemia you cannot have reperfusion injury. Ischemia is defined as inadequate energy supply to meet the current demands, that is, an energy supply/demand ratio of less than 1; normally, the O2 supply/demand ratio is >1. Since the adenosine triphosphate (ATP) necessary to support the energetic needs of the heart is provided predominantly by oxidative phosphorylation, with scant generation of high-energy phosphates through anaerobic metabolism, the heart is nearly an obligate aerobic tissue. Therefore, the energy consumption of the heart can be approximated by the oxygen consumption. The limited availability of anaerobic energy-generating pathways mandates that O2 be in constant supply. A very brief discussion of the determinants of O2 supply and demand follows.
Oxygen supply: Ischemia may be caused by either a decrease in O2 supply relative to demands (supply ischemia) or an increase in O2 demands relative to supply (demand ischemia). O2 supply is determined by the O2 extraction (arterial-coronary venous O2 difference) and coronary blood flow. Since O2 extraction by the heart is normally ˜75%, and its physiologic limit is approximately 95%, only limited amounts of oxygen may be obtained through additional extraction. The majority of O2 is therefore supplied by adjustments in coronary blood flow, which under normal conditions may increase 4- to 5-fold to meet demands. This O2 is largely present as hemoglobin-bound, with only a minor fraction (˜1.5%) dissolved in plasma. This limited dissolved O2 will become important when we discuss crystalloid cardioplegia solutions in which oxygen availability is limited to that dissolved in solution.
Oxygen demand: Overall myocardial oxygen demand of the normal working heart is determined by the work of the entire heart. The majority of this overall O2 consumption is attributed to utilization by the left ventricle. In a nutshell, the determinants of left ventricular O2 demand include pressure-volume work or wall stress, heart rate, temperature, inotropic state, basal metabolism, and ionic homeostatic mechanisms (ATP-dependent pumps) required to re-equilibrate ionic balance after electromechanical activity. In the immediate postoperative heart, energy demands may be related to oxidative energy diverted to myocyte repair.
The energy related to maintain ionic equilibrium (˜5% of total demand) is an ongoing demand even when the heart is not contracting since ATP-dependent pumps are constantly adjusting calcium and sodium influx, as well as potassium efflux. This energy demand of ionic equilibration is important in determining ongoing oxygen demands in the arrested heart, as discussed below. How oxygen demands are changed by hypothermia is discussed later.
Severity and Duration of Ischemia in Cardiac Surgery
The potential for myocardial injury is related, in part, to the duration of ischemia. The time to onset of irreversible ischemic injury will depend on many factors, including the severity of ischemia, myocardial temperature, ambient energy demands, and collateral blood flow. However, irreversible injury can become apparent after as little as 30 to 45 minutes of coronary occlusion in the working myocardium. However, shorter durations of global ischemia can result in mild to severe systolic and diastolic dysfunction without irreversible tissue necrosis (left ventricular “stunning” (33)). Therefore, the duration of ischemia, both preceding CPB and intraoperatively after application of the cross-clamp, has often been used as a predictor of postoperative myocardial injury in experimental models, and is one of the underlying considerations in the correlation between total ischemic time (CPB, cardioplegia) and clinical outcomes. Because the surgical team often does not have control over the duration of warm ischemia (particularly antecedent ischemia caused by coronary occlusive disease), other aspects of elective ischemic time are more often targeted for modification to reduce the severity of ischemia and hence postischemic injury. Therefore, strategies such as (1) initiating rapid asystole, (2) venting the left ventricle to reduce consequences of chamber distension resulting in increased wall stress, and (3) imposing cardiac hypothermia all specifically target the reduction of myocardial energy/oxygen demands, thereby increasing the limits of “safe” ischemic time during aortic cross-clamping. With these cardioprotective strategies in place, the duration of aortic clamping that can be safely imposed can be increased from as little as 15 to 45 minutes to several hours using hypothermic cardioplegia (Fig. 9.2). Reducing to almost zero the determinants of myocardial work and oxygen demand is the sine qua non of myocardial protection. In the section on future directions, eliminating ischemic conditions without additional oxygen-related injury and changing the milieu more gradually will be discussed.
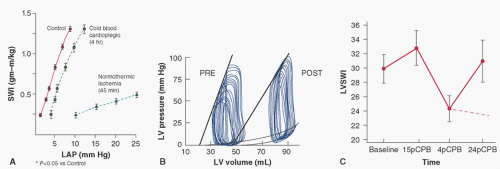 FIGURE 9.2. Left ventricular performance measured by in situ Starling curves (A) or end-systolic pressure-volume relations (conductance catheter) (B) after 45 minutes of normothermic unprotected global ischemia and reperfusion. C: Left ventricular stroke work index in patients before (Baseline) or after (pCPB) 15 minutes, or 4 and 24 hours postcardiopulmonary bypass (CPB). The dashed line between 4 and 24 hours after discontinuation of CPB represents further decreases in cardiac performance secondary to onset of irreversible injury. (A, adapted from Rosenkranz ER, Buckberg GD. Myocardial protection during surgical coronary reperfusion. J Am Coll Cardiol 1983;1:1235-1246, with permission.) |
The determinants of surgical ischemic injury can be summarized as:
The duration and severity of antecedent” and “unprotected” ischemia: Although it has been recognized for decades that ischemia time (duration) impacts tissue injury and postischemic salvage and function, an increasing appreciation for the caveat that the manner in which the tissue is reperfused significantly alters the outcome. It is now increasingly understood that reperfusion injury can occur
by merely eliminating ischemic conditions abruptly or in the presence of an inappropriate oxygen gradient. In the case of the imposition of aortic cross-clamping, there is total elimination of native flow with the exception of that supplied by noncoronary collateral flow. Global contractile function may be impaired before necrosis or apoptosis are evident (“stunning”). It is the reduction of myocardial oxygen demand that protects the heart in this circumstance.
The elimination of electromechanical activity during cross-clamping is paramount because its mechanical activity increases O2 demands even in the presence of hypothermia (discussed further in the section on Hypothermia).
Myocardial temperature: Temperature influences metabolism by the “Q10 effect,” in which the tissue metabolic rate decreases by half for each 10°C decrease in temperature; conversely, myocardial metabolism doubles when the heart rewarms by 10°C. Hypothermia can buy biologic time during ischemia.
The nutritional and stress status of the heart: For example, catecholamines increase the O2 demands by increasing contractility and exerting an O2 wasting effect.
Presentation of comorbidities such as hyperlipidemia which increase the vulnerability to ischemia-reperfusion injury for any given duration of ischemia. This effect is independent of energy demands, but is influenced more by mechanisms exerted at reperfusion, that is, the capacity to generate O2 radicals, a pro-inflammatory state of the endothelium and inflammatory cells, and calcium handling by the tissue.
Determinants of Reperfusion Injury
Although reperfusion of the myocardium is the ultimate goal of both surgical and nonsurgical revascularization, abrupt reperfusion carries with it the potential for extending postischemic injury to cardiomyocytes, coronary vascular endothelium, and the microvasculature. Indeed, reperfusion injury associated with rapid changes in circulatory conditions may be responsible for ˜50% (34) or more of the ultimate infarct size, and is an important contributor to postsurgical mortality and morbidity as well (29). We can define surgical reperfusion injury as an incremental increase in the pathology that is observed after ischemic injury alone. Reperfusion injury extends or accelerates damage from that observed during ischemia alone, and occurs after the onset of reperfusion (i.e., cardioplegia) or reperfusion (i.e., cross-clamp removal). Viewed another way, reperfusion injury can occur at the instant of the elimination of ischemic conditions with reestablishment of flow. Hence, reperfusion injury can be viewed as representing an increment of injury that had begun during ischemia but progresses or manifests during the reperfusion phase. Other investigators have described de novo injury starting at reflow or, in a newer construct, at the time of the elimination of ischemia (even if that is accomplished by a reduction in oxygen demand). Reperfusion injury can be categorized as either reversible or irreversible. (1) Reversible injury includes temporary contractile dysfunction (“stunning”) in the absence of morphologic injury or necrosis, or (2) irreversible, which includes necrosis and apoptosis. Biomarker enzymes such as creatine kinase (CK) or cardiac troponin T (cTnT) or I (cTnI) released by disrupted cells have been observed to be elevated during reperfusion rather than during ischemia. In the past, debate has often been spirited regarding whether reperfusion injury even exists (35,36,37,38) and whether reperfusion injury kills cells that are salvageable at the end of ischemia. However, a consensus has developed in both the surgical, cardiology, and cardiovascular research communities that reperfusion is indeed an important component of postischemic cardiac injury in animal models (39) and in humans (34,40) and that alterations in reperfusion strategies can mitigate injury.
Among the more important mechanisms in the pathophysiology of myocardial ischemia-reperfusion injury are (1) the duration and severity of antecedent ischemia, (2) oxygen radicals generated not only by neutrophils but also by myocytes and vascular endothelium, (3) sodium (Na+) and Ca2+ influx and loss of normal intracellular Ca2+ homeostasis, (4) neutrophils and neutrophil-derived products that constitute a generalized inflammatory response to reperfusion, and (5) mitochondrial dysfunction and opening of the mitochondrial permeability transition pore (mPTP). Interestingly, the presence of hyperoxic or even relatively hyperoxic conditions can ramp up or trigger all of these mechanisms. As such, these mechanisms do not operate independently, nor are they mutually exclusive. Importantly, reperfusion injury is a malleable process that can be attenuated by utilizing a number of strategies targeting these various perpetrators of injury. In the operating room during cardiac surgery, and more and more frequently in the modern era in the interventional lab, various forms of circulatory support techniques and cardioprotective drugs are employed. The realization that circulatory support can manipulate reperfusion therapy by altering oxygen supply/demand relationships is critical for any operator that employs it. Such pumps provide an avenue for both the reduction of oxygen demand and for the delivery of therapeutic agents via the pump, which can be administered intravenously or by using cardioplegia as a vehicle. Using cardioplegia offers a unique opportunity to modify the conditions of reperfusion (hydrodynamics, temperature, route of delivery) and the composition of the solution (blood, crystalloid, pH, metabolic substrates, hypocalcemia, oxygen, pharmaceuticals) that target the pathophysiologic mechanisms of injury. It is now also recognized that the initiation of circulatory support in it itself offers an opportunity to mitigate injury by paying attention to conditions at the moment support is begun and ischemic conditions are eliminated.
Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS)
Molecular oxygen is relatively inert by virtue of the shared electron pair in the outer molecular shell. However, oxygen
must be viewed as a pharmaceutical and potentially toxic agent in cardiovascular medicine and surgery. ROS are radicals or unstable oxygen molecules derived from oxygen. Important oxygen radicals are superoxide anion (–O2) and hydroxyl anion (•OH).OH). However, other oxygen-related molecules that are not technically radicals are considered biologically reactive nonetheless. An example of this would be hydrogen peroxide (H2O2). In addition, hypochlorous acid (HOCl) and chloramines derived from neutrophils exert potent oxidative injury to biologic tissues.
must be viewed as a pharmaceutical and potentially toxic agent in cardiovascular medicine and surgery. ROS are radicals or unstable oxygen molecules derived from oxygen. Important oxygen radicals are superoxide anion (–O2) and hydroxyl anion (•OH).OH). However, other oxygen-related molecules that are not technically radicals are considered biologically reactive nonetheless. An example of this would be hydrogen peroxide (H2O2). In addition, hypochlorous acid (HOCl) and chloramines derived from neutrophils exert potent oxidative injury to biologic tissues.
Free-radical molecules that are derived from nitrogen are termed RNS and include nitric oxide (NO•), peroxynitrite (ONOO-), nitrogen dioxide (NO2•) and nitrosyl hydride (HNO). In vascular endothelium, NO• is constitutively generated by endothelial nitric oxide synthase (eNOS or NOS-3). eNOS activity is dependent on Ca2+-calmodulin, flavin, NADPH, and tetrahydrobiopterin (BH4). NO• is generated by an electron from the guanadino nitrogen of L-arginine to incorporate oxygen to generate NO• and the byproduct L-citrulline. NO• has an extremely short half-life (seconds) in vivo, and binds tightly to hemoglobin. OONO- is formed by a nonenzymatic biradical reaction between NO• and –O2. NO• has been linked to biologic signaling in cardioprotection, particularly anti-inflammatory effects, endothelial function, vasorelaxation (hence its early name as endothelial-derived relaxing factor, EDRF), neurotransmission, immune regulation and defense mechanisms.
NO• homeostasis is key in normal regulation of blood flow and responses to stress, and in preventing endothelial dysfunction. NO• homeostasis is a balance between NO• generation, bioavailability, and degradation or quenching by oxidants such as –O2. NO• has pleiotropic effects in the cardiovascular system by paracrine effects: (1) NO• is a physiologic regulator of blood pressure and flow, and counteracts local and systemic vasoconstrictors such as endothelin (ET-1); (2) it inhibits neutrophil adhesion to vascular endothelium; (3) it inhibits platelet aggregation; (4) it reduces myocardial oxygen consumption (41); (5) it has positive lusitropic and inotropic effects; (6) it scavenges –O2 and therefore is directly antioxidant; and (7) it is anti-proliferative. The effects on oxygen consumption and inotropy are somewhat controversial (42,43). The role of NO• in ischemia-reperfusion injury and cardioprotection is discussed in more detail under the section on endothelial dysfunction and NO• donors.
Reactive Oxygen Species
The oxygen radical is highly reactive with a broad range of biologic substances including sugars, amino acids, phospholipids, and DNA. With such a large catalog of biologic targets, ROS and their metabolites are potentially toxic to cells, and at the same time they are critical in the host defense mechanism and bactericidal activities of neutrophils and phagocytes (44) and participate in cardioprotective signaling. In this dualistic role of ROS as good guy-bad guy, it is proposed that low-level production of ROS, particularly from mitochondria, acts as a signal for cardioprotective conditioning responses (preconditioning, postconditioning) and stimulation of cardioprotective kinase pathways, while large-scale generation of ROS from inflammatory cells or dysfunctional mitochondria is deleterious. In the ischemic-reperfused myocardium, ROS-induced injury includes peroxidation of lipid components of cellular membranes leading to damage to sarcolemmal cell membrane and other membranous organelles such as mitochondria and sarcoplasmic reticulum (45). ROS also cause the mPTP to open when generated in an environment of Ca2+ accumulation and normalized pH (conditions achieved during reperfusion), which leads to a collapse of the membrane potential, loss of metabolic capacity, and release of pro-apoptotic factors. This is discussed in more detail below under the role of mitochondria in reperfusion injury. The vascular endothelium is particularly vulnerable to ROS-mediated injury, which manifests as impairment of vascular endothelial function through attenuated production of vasoactive and anti-inflammatory autacoids such as adenosine (46,47,48) and NO• (49), and increased production of pro-inflammatory and vasoconstrictive molecules. These injury mechanisms contribute significantly to postischemic dysfunction, dysrhythmias, and morphologic injury such as necrosis and apoptosis.
Sources of Oxygen Radicals
Both enzymatic and nonenzymatic reactions produce oxygen radicals. The major reactions and their sources are shown in Figure 9.3. ROS are generated by a number of sources, including activated neutrophils, cardiac myocytes, mitochondria, and the vascular endothelium.
Neutrophils: Activated neutrophils represent the major source of ROS in the heart under stress, including –O2, •OH, and HOCl species. Neutrophils are activated by various chemotactic factors stimulated by either CPB or ischemia-reperfusion, including the complement anaphylatoxins C3a and C5a, f-Met-Leu-Phe, tumor necrosis factor alpha (TNFα), interleukin-8 (IL-8), and platelet-activating factor. These factors trigger a “respiratory burst” of ROS. CPB (independent of ischemia) activates the complement cascade (50,51), with the subsequent activation and adherence of neutrophils (52) to the vascular endothelium primarily, which subsequently triggers the release of cytotoxic products, all of which contribute to tissue injury in the ischemic-reperfused myocardium (52,53). During the respiratory burst that occurs seconds after stimulation by cytokines or ischemia-reperfusion, the –O2 in the neutrophil is produced by the membrane-associated NADP oxidase, which transfers an electron to molecular oxygen (Fig. 9.3, reviewed in Sheppard et al. (54)). Production of H2O2 by neutrophils or the reduction of –O2 to H2O2 by superoxide dismutase (SOD) provides substrate for generation of •OH, or the oxidant HOCl by myeloperoxidase. HOCl reacts with low molecular weight amines to give rise to lipophilic chloramines, which can promote membrane lipid peroxidation.
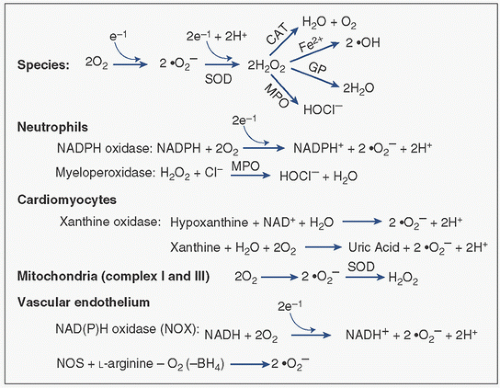 FIGURE 9.3. The species and sources of reactive oxygen species (ROS) generated by neutrophils, cardiomyocytes and their mitochondria, and vascular endothelium. SOD, superoxide dismutase; CAT, catalase; GP, glutathione peroxidase; MPO, myeloperoxidase; HOCl, hypochlorous acid; e-1, electron; NOX, NAD(P)H oxidase. |
Cardiomyocytes: Xanthine oxidase (XO) is a major source of ROS from cardiomyocytes. Normally, XO exists mostly in the dehydrogenase form and uses nicotinamide adenine dinucleotide (NAD) as an electron acceptor. Xanthine dehydrogenase is converted to the oxidant-generating enzyme XO during ischemia. Both hypoxanthine and xanthine accumulate during ischemia. Hypoxanthine is converted to xanthine by XO, which is in turn converted to uric acid by the same enzyme, which forms –O2. During ischemia, however, two events favor the xanthine oxidase reaction: (1) hypoxanthine accumulates as a result of the sequential catabolism of purine high-energy phosphates (ATP, ADP, AMP) and deamination of adenosine, and (2) the dehydrogenase form of the enzyme is converted to the superoxide-producing oxidase form by a protease activated by increased levels of intracellular calcium (55). XO is reported to be localized to the vascular endothelium in substantial quantities (56). However, the importance of XO to the pathologic generation of ROS in humans remains controversial (57).
Mitochondria: Mitochondria are a major noninflammatory site of ROS generation (58). ROS are generated at complex I (NADH/ubiquinone oxidoreductase) and complex III (ubiquinol/cytochrome c oxidoreductase). The latter site catalyzes the conversion of O2 to –O2• by a single-electron transfer. –O2 can be converted in the mitochondrial matrix to H2O2 by matrix superoxide dismutase. Since mitochondria do not have catalase, the H2O2 can be degraded by nonenzymatic reaction with glutathione (GSH) by glutathione peroxidase or by enzymatic reaction with thioredoxin reductase. The ROS species target cysteine residues on the protein moieties of the mitochondrial membrane and polyunsaturated fatty acids, and cause intramolecular cross-linkages and protein aggregates. The •OH causes peroxidation of membrane lipids. ROS induce Ca2+ release from the mitochondria, which may cause an imbalance in Ca2+ handling and stimulation of Ca2+-dependent proteases, nucleases and phosphatases leading to functional and morphologic damage to the cell.
Vascular endothelium: Superoxide anion is produced by vascular endothelium by XO as described above for cardiomyocytes (Fig. 9.3). A more important source of –O2 from the vascular endothelium may be the NAD(P)H (reduced NAD phosphate) oxidase system. This enzyme is a membrane-bound, flavin-containing NADH/NADPH-dependent oxidase present in endothelial and vascular smooth-muscle cells. The molecular structure, function, and regulation are somewhat similar to those of the neutrophil enzyme system, requiring assembly of cytosolic components onto the membrane components before –O2 generation can occur. However, –O2 production in the presence of NO• produced by endothelium favors the biradical neutralization and elimination of NO• and the formation of peroxynitrite (ONOO-). The elimination of NO• impairs not only vascular relaxation and autoregulatory control of postischemic myocardial perfusion, but also fails to inhibit neutrophil-mediated postischemic inflammatory responses.
Other sources of ROS include the oxidation of catecholamines, metabolism of arachidonic acid to peroxy compounds, and generation of •OH by the cyclooxygenase and lipoxygenase pathways, and by the metal-catalyzed Haber-Weiss reactions. The latter series of reactions may be important in the setting of CPB because they can be recruited by neutrophil activation during CPB (59). In addition, iron-containing products released by hemolysis may facilitate this reaction.
When are Oxygen Radicals Generated?
Myocardial ischemia favors the generation of ROS. However, the myocardium has a cadre of the endogenous antioxidants superoxide dismutase, catalase, reduced glutathione (GSH), glutathione peroxidase, glutathione reductase. Normally, upregulation of these antioxidants and phase 2 enzymes reduces oxidant-mediated injury (60), but they are depleted or can be overwhelmed during ischemia, thereby attenuating the natural defense mechanisms of the heart (61). Therefore, ischemia creates the biochemical setting for oxyradical production and tissue vulnerability to ROS-mediated damage. However, the primary substrate, oxygen, is in limited supply during ischemia, and is not readily available until reperfusion. Oxygen radicals are not seen in great abundance until the onset of reperfusion. A clinical study by Prasad et al. (62) show that oxygen radicals are released during and 24 hours after CPB in CABG patients. Indirect evidence to support this reperfusion oxidative burst comes from a wealth of data in which inhibitors or scavengers of oxygen radicals, or anti-neutrophil agents, exerted beneficial effects when administered during reperfusion.
In the surgical setting, the myocardium may be vulnerable to ROS-induced injury at several points when oxygen is introduced into the myocardium: (1) the initial delivery of cardioplegia into the heart with antecedent unprotected ischemia, or initial delivery of oxygenated cardioplegia to a newly revascularized myocardial segment; (2) intermittent reinfusions of cardioplegia if a multidose strategy is being used; and (3) release of the aortic cross-clamp to initiate reperfusion after the cardioplegia phase. The quantity of ROS generated during cardiac surgery depends on the severity of the preceding ischemic injury, activation and recruitment of neutrophils, the level of oxygen in cardioplegic solutions or blood oxygenated by the extracorporeal circuit, and the status of endogenous scavengers and inhibitors potentially depleted during ischemia. Blood cardioplegia theoretically provides a greater oxygen substrate for oxygen radical production than do crystalloid solutions. However, plasma-dissolved oxygen may be the pool from which oxygen is drawn for the generation of oxygen radicals, and the oxygen content in this compartment may not differ dramatically between blood and crystalloid solutions. The oxygen in blood cardioplegia may be reduced with “normoxic” or even “deoxygenated” blood cardioplegia (63) or during reperfusion (64,65). In addition, blood cardioplegia may offer the advantage of endogenous antioxidants (superoxide dismutase, catalase, glutathione, and albumin) that are not present in crystalloid cardioplegia solutions.
Intracellular Na+ and Ca2+ Dyshomeostasis
Under normal physiologic conditions, intracellular Ca2+ concentrations are maintained at low concentrations (<200 nmol) against a 5,000-fold trans-sarcolemmal gradient by voltage-gated Ca2+ channels that remain closed until activation during phase 2 of the action potential. The regulation of intracellular and intramitochondrial Ca2+ is tightly linked to regulation of intracellular Na+. Intracellular Na+ concentration is normally controlled by the ATP-dependent Na+/K+ pump which moves Na+ outward and K+ inward against their respective concentration gradients, and the energy-independent Na+/H+ exchanger which normally removes one H+ from the cytosol in exchange for one Na+ moving into the cytosol. Further Na+ accumulation mainly occurs through the Na+/Ca2+ exchanger which normally operates in the forward direction during diastole to extrude intracellular Ca2+ in exchange for influx of Na+ into the cytosol.
In ischemic-reperfusion myocardium, Ca2+ influx occurs through multiple pathways: (1) diffusion through overt membrane disruptions or opened Ca2+ channels, fueled by the differential concentration gradient; (2) reversal or inhibition of the Na+/Ca2+ exchanger driven by the intracellular accumulation of Na+, (3) attenuated cyclical Ca2+ uptake and release from the sarcoplasmic reticulum and mitochondria, and (4) facilitation of entry via α-adrenoreceptor activation and cyclic adenosine monophosphate (AMP)-dependent processes, which increase during postischemic catecholamine release.
The accumulation of intracellular Na+ and Ca2+ during ischemia-reperfusion are linked events leading to Ca2+ overload which stimulates Ca2+-dependent proteases that damage membranes and ion transport mechanisms at the moment of re-exposure to molecular oxygen, opening of the mPTP, depletion of high-energy phosphate stores, the development of local or global contracture (“stone heart”), and triggers the transition to cell death through both apoptosis and necrosis. The balanced Ca2+ transport systems are potentially injured by exposure to oxygen radicals, cytokines, and activated complement during CPB and reperfusion. Na+ and Ca2+ dyshomeostasis occurs to differing degrees during ischemia and during reperfusion, as described below.
Ischemia
During ischemia, glycolysis generates an abundance of H+ and only a limited amount of ATP (2 moles/mole glucose) compared to oxidative phosphorylation (36 moles ATP/mole glucose). The accumulation of intracellular H+ stimulates activity of the Na+/H+ exchanger (NHE1) in favor of H+ efflux and Na+ influx; at this time, the reduced availability of ATP decreases Na+/K+ ATPase activity. Therefore, both the attenuated Na+/K+ pump activity and increased activity of the Na+/H+ exchanger result in intracellular Na+ accumulation. Additional Na+ may
enter the cell through sarcolemmal nonactivating Na+ channels, for example, the “window currents” generated during ischemia-induced cellular depolarization. However, some evidence suggests that the accumulation of H+ in both the extracellular and intracellular spaces attenuates the H+ gradient, thereby reducing but not eliminating the participation of the Na+/H+ exchange mechanism in intracellular Na+ accumulation during ischemia (66). However, the accumulation of Na+ in the cytosol during ischemia would favor reversal of the energy-independent Na+/Ca2+ exchanger in favor of extruding Na+ in exchange for Ca2+ influx, thereby favoring its accumulation in the cytosol. Entry of Ca2+ into the mitochondria through the Ca2+ uniporter depends on the Ca2+ electrochemical gradient and the presence of a mitochondrial transmembrane potential. Ca2+ enters the mitochondria during early ischemia, but further increases and overload are prevented by dissipation of the mitochondrial transmembrane potential (67). In addition, the unregulated activation of Ca2+-dependent protease enzymes such as calpains is inhibited by intracellular acidosis, and is delayed until intracellular pH is renormalized during reperfusion (68). Therefore, Ca2+ accumulation and Ca2+-mediated damage occurs to a greater extent during reperfusion (69,70,71) as discussed below.
enter the cell through sarcolemmal nonactivating Na+ channels, for example, the “window currents” generated during ischemia-induced cellular depolarization. However, some evidence suggests that the accumulation of H+ in both the extracellular and intracellular spaces attenuates the H+ gradient, thereby reducing but not eliminating the participation of the Na+/H+ exchange mechanism in intracellular Na+ accumulation during ischemia (66). However, the accumulation of Na+ in the cytosol during ischemia would favor reversal of the energy-independent Na+/Ca2+ exchanger in favor of extruding Na+ in exchange for Ca2+ influx, thereby favoring its accumulation in the cytosol. Entry of Ca2+ into the mitochondria through the Ca2+ uniporter depends on the Ca2+ electrochemical gradient and the presence of a mitochondrial transmembrane potential. Ca2+ enters the mitochondria during early ischemia, but further increases and overload are prevented by dissipation of the mitochondrial transmembrane potential (67). In addition, the unregulated activation of Ca2+-dependent protease enzymes such as calpains is inhibited by intracellular acidosis, and is delayed until intracellular pH is renormalized during reperfusion (68). Therefore, Ca2+ accumulation and Ca2+-mediated damage occurs to a greater extent during reperfusion (69,70,71) as discussed below.
Reperfusion
Na+ and Ca2+ dyshomeostasis during reperfusion is a significant factor contributing to postischemic myocardial injury and functional impairment (72,73). The ion dyshomeostasis described above sets the stage for explosive reperfusion/reoxygenation damage. It is increasingly recognized that abrupt re-exposure to oxygen itself is the trigger for damage. Excessive gradients of perfusate-to-tissue oxygen tension can be mitigated. In addition, the events leading to abnormal Ca2+ handling which determine cell survival versus cell death are as follows:
Rapid renormalization of intracellular pH: Reperfusion washes out lactate and re-establishes the intracellular-extracellular H+ gradient (extracellular < intracellular) which allows the forward motion of the Na+/H+ exchanger favoring removal of H+ (renormalization of intracellular pH) and subsequent intracellular accumulation of Na+; other factors contributing to normalization of intracellular pH are activation of lactate/H+ co-transport mechanism and activation of the NaHCO3 co-transporter.
Renormalization of intracellular Na+: During the initial phase of reperfusion, ATP is regenerated, and if the Na+/K+ AT-Pase is not damaged by proteolytic actions of calpain enzymes, intracellular Na+ levels are renormalized and the linked Na+/H+ exchange—Na+/Ca2+ exchange systems will not be activated. Otherwise, if the Na+/K+ ATPase pump is damaged, intracellular Na+ accumulation will engage the Na+/Ca2+ exchange mechanism with net intracellular accumulation of Ca2+.
Ca2+ uptake and release by the sarcoplasmic reticulum (SR): The SR is the main Ca2+ storage site of the cardiomyocytes. Ca2+ in the micro-domain proximate to the closely co-localized SR and interfibrillar mitochondria is cyclically taken up by the SR (lowering of cytosolic concentration) during diastole, and Ca2+ is released into that micro-domain by the ryanodine receptors (RyR) during systole. During both unprotected and protected ischemia, sequestration of Ca2+ into the sarcoplasmic reticulum competes for a limited amount of ATP. This paradox creates a cycle whereby a high intracellular Ca2+ level binds high-energy phosphates to further limit ATP availability, which then attenuates the ability of the SR to remove the Ca2+ actively from the cytoplasm, resulting in a further increase in cytosolic Ca2+. If other Ca2+ handling mechanisms are not engaged, the release of Ca2+ from the SR RyR may lead to overload. This activity of the SR mechanism may be related to the severity of ischemia, being less so during short periods of ischemia.
So, what events in the balance of Ca2+ regulation determines if a cell lives or dies during reperfusion? Some evidence suggests that the timing of normalization of pH and Ca2+ handling mechanisms are important. If pH normalization occurs before Ca2+ handling mechanisms are restored, then Ca2+ overload, in conjunction with ROS generation and a normalized intracellular pH environment, will trigger opening of the mPTP causing cell death. However, if Ca2+ handling mechanisms are restored before pH is normalized, for example, by maintaining tissue acidosis during early reperfusion, then Ca2+ overload can be avoided. Interestingly, venous blood-based perfusate has been preliminarily observed to mitigate both over-oxygenation during early reperfusion and delayed normalization of pH, thereby creating a “permissive acidemia.”
The Inflammatory and Innate Immunity Responses to Ischemia-Reperfusion and Cardiopulmonary Bypass
Injury mechanisms of ischemia-reperfusion and CPB involve inflammatory and innate immune responses. Cardiomyocytes generate pro-inflammatory cytokines, chemokines and adhesion molecules that direct the emigration of inflammatory cells into ischemic-reperfused tissue regions and amplify the inflammatory response. The following sections will describe the inflammatory process involving neutrophils, pro-inflammatory cytokines, and complement, and the innate immune response involving toll-like receptors (TLRs).
Neutrophils and Neutrophil-Mediated Injury
Neutrophils play a critical role in the pathogenesis of myocardial reperfusion injury (74,75,76). The inflammatory component of reperfusion injury requires an interaction between activated neutrophils and vascular endothelium during the early moments of reperfusion, which is prerequisite to activation of the full local inflammatory cascade. A well-orchestrated sequence of events mediates the interaction between adhesion molecules on neutrophils and endothelium. These adhesion molecules are categorized into three families:
The selectins (P-selectin, L-selectin, E-selectin) are glycoproteins involved in the interactions between neutrophils
and the vascular endothelium in the early moments of reperfusion (77,78,79). P-selectin is stored in Weibel-Palade bodies in endothelial cells, and its surface expression is triggered by pro-inflammatory mediators such as oxygen radicals, thrombin, complement components, cytokines, histamine, and H2O2 released during ischemia-reperfusion and CPB. IL-8 released from hypoxic endothelial cells also increases the adhesiveness of endothelial cells for neutrophils (80). P-selectin surface expression peaks after 10 to 20 minutes of reperfusion, and P-selectin is subsequently shed to soluble fragments in blood. In contrast to P-selectin, L-selectin is constitutively expressed on the surface of neutrophils and may be the counterligand for P-selectin during early reperfusion (81). Loose adherence and “rolling” of neutrophils on endothelium involves interaction with a glycoprotein sialyl Lewisx or a high-affinity glycoprotein ligand termed P-selectin glycoprotein ligand-1 (PSGL-1) (82). The third member of the selectin family, E-selectin, is expressed on the surface of endothelial cells. It is expressed later in reperfusion (4-6 hours) and may therefore be involved in later events, such as infarct extension.
The β2-integrins (CD11/CD18 complex) are a family of glycoproteins located on external membranes of neutrophils. There are three distinct α-chains (CD11a, CD11b, CD11c) and a common β-subunit. Surface expression of perhaps the major complex CD11b/CD18 is triggered by a number of pro-inflammatory mediators, including circulating and endothelium-derived (acylated) platelet-activating factor. After initial tethering of neutrophils by endothelial P-selectin, firm adherence is mediated by interaction with CD11b/CD18 and its counterligand ICAM-1 on the endothelium.
The third family of adhesion molecules is the immunoglobulin superfamily, including ICAM-1 (intercellular adhesion molecule-1), VCAM-1 (vascular cell adhesion molecule-1), and PECAM-1 (platelet-endothelial cell adhesion molecule-1). ICAM-1, the counterligand for CD11/CD18 on neutrophils, is constitutively expressed on endothelial cells and is upregulated by cytokines 2 to 4 hours after myocardial ischemia-reperfusion (83), coincident with the upregulation of CD11/CD18. Subsequently, a high-affinity bond between the neutrophil and endothelial cell occurs via integrins (CD11/CD18) on neutrophils and ICAM-1 and ICAM-2 on endothelial cells. Once the neutrophil has firmly attached to the endothelial surface, it migrates through the endothelial cell junctions to the myocardium, partly mediated by IL-8. The shedding of L-selectin from the neutrophil and the presence of PECAM on the endothelium may be required for neutrophil transendothelial migration. The prerequisite nature of this interaction between neutrophils and adhesion molecules makes anti-neutrophil and anti-adhesion therapy a potentially effective strategy, as these agents intervene at proximal points in the cascade (discussed below).
Evidence supporting the rapid accumulation of neutrophils within ischemic-reperfused myocardium following the neutrophil-endothelial cell interactions was appreciated not only by our laboratory, but also by Sommers and Jennings (84). We (85,86,87,88,89) and others (90) have demonstrated an association between the extent of postischemic injury and neutrophil accumulation within ischemic-reperfused myocardium as well as a reduction of postischemic injury associated with neutrophil suppression. As shown in Figure 9.4, Lefer et al. (91) demonstrated that neutrophil adherence to coronary artery endothelium occurs in advance of accumulation in extravascular sites, representing the lag time involved in transmigration of intravascular neutrophils. In addition, neutrophil accumulation occurs in advance of the development of necrosis, consistent with active neutrophil participation in cell demise. Brix-Christensen et al. (92) reported that neutrophils accumulated in the hearts and other organs of neonatal pigs placed on CPB. Therefore, neutrophil accumulation occurs in both postischemic and post-CPB hearts.
The importance of neutrophils in postbypass reperfusion injury has been well-established in both animal and human models. The myocardium is exposed to neutrophils during the delivery of intermittent or continuous blood cardioplegia, or during reperfusion (after cross-clamp removal) following either crystalloid or blood cardioplegia. Schwartz et al. (93) showed that CPB circuitry directly “primes” neutrophils by depositing C3b on extracorporeal tubing, leading to neutrophil activation and sequestration. Further evidence for the activation of neutrophils following CPB was suggested by the finding of increased neutrophil surface expression of CD11b/CD18 and decreased surface expression of L-selectin on cells circulated in simulated CPB circuits (94,95). Finn et al. (96) noted in patients undergoing CPB that a rise in circulating IL-8 begins at reperfusion with rewarming of the patient after systemic hypothermia, and closely correlates with the rise in neutrophil count and circulating elastase. Similarly, Schwartz et al. (93) and Gessler et al. (97) found higher levels of the pro-inflammatory mediators soluble IL-6 and IL-8 6 hours after bypass than before bypass.
The time course for the upregulation of neutrophils and various adhesion molecules is equally important in designing treatment strategies for patients undergoing extracorporeal bypass. In a clinical study, Galiñanes et al. (98) investigated the perioperative temporal changes in the expression of various neutrophil surface adhesion molecules, concluding that downregulation of PECAM-1 is an early indicator of neutrophil activation and may therefore represent a target for therapy aimed at reducing the inflammatory response associated with CPB.
The role of temperature in the activation of neutrophils or other inflammatory mediators following CPB in humans has been investigated. Hypothermia has been reported to attenuate expression of adhesion molecules in some models of tissue injury (99,100). Some investigators have reported minimal
or no difference in neutrophil activation and expression of neutrophil adhesion molecules between normothermic (34°C-37°C) and hypothermic (28°C-30°C) CPB (101,102), whereas others note an attenuation in the expression of IL-8 and neutrophil elastase activity during normothermic CPBsurgery (103). Hypothermia has been shown to delay, but not prevent, the increased expression of adhesion molecules such as CD11b and CD11c (101). When measured 4 hours after bypass, the levels of IL-1α, soluble ICAM-1 (sICAM-1), and elastase are higher after normothermic CPB than after hypothermic CPB (104,105). In conclusion, further research is required to determine the most appropriate temperature of cardioplegic administration to minimize the deleterious consequences of neutrophil-mediated damage. Interventions designed to attenuate neutrophil-related myocardial injury following CPB will be discussed later.
or no difference in neutrophil activation and expression of neutrophil adhesion molecules between normothermic (34°C-37°C) and hypothermic (28°C-30°C) CPB (101,102), whereas others note an attenuation in the expression of IL-8 and neutrophil elastase activity during normothermic CPBsurgery (103). Hypothermia has been shown to delay, but not prevent, the increased expression of adhesion molecules such as CD11b and CD11c (101). When measured 4 hours after bypass, the levels of IL-1α, soluble ICAM-1 (sICAM-1), and elastase are higher after normothermic CPB than after hypothermic CPB (104,105). In conclusion, further research is required to determine the most appropriate temperature of cardioplegic administration to minimize the deleterious consequences of neutrophil-mediated damage. Interventions designed to attenuate neutrophil-related myocardial injury following CPB will be discussed later.
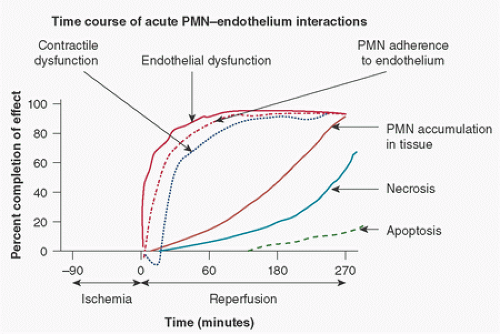 FIGURE 9.4. The postischemic time course of endothelial injury, contractile dysfunction, neutrophil (PMN) adherence and accumulation in tissue, necrosis, and apoptosis. These events are indicated on the y-axis as a percent completion of that event (0% not started, 100% is complete). Note that PMN adherence parallels that of endothelial dysfunction representing PMN-mediated damage. Note also that accumulation in tissue lags behind adherence representing emigration time. (Data obtained, in part, from Zhao ZQ, Nakamura M, Wang NP, et al. Dynamic progression of contractile and endothelial dysfunction and infarct extension in the late phase of reperfusion. J Surg Res 2000;94:133-144.) Apoptosis is a slower process than necrosis as observed by Zhao et al. (565). (Adapted in part from Lefer AM, Tsao PS, Lefer DJ, et al. Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. FASEB J 1991;5:2029-2034.) |
Complement in CPB and Myocardial Reperfusion Injury
The complement cascade, particularly the alternative pathway stimulated by contact activation, is activated independently by myocardial ischemia-reperfusion and CPB, and contributes importantly to the pathologic sequelae of CPB (106). Complement fragments such as the anaphylatoxins C3a and C5a are generated and released both systemically and locally by the ischemic-reperfused myocardium (107). The activation and migration of neutrophils to ischemic-reperfused myocardium requires stimulation by chemotactic factors, most notably complement fragments (C3a and C5a), eicosanoid products such as leukotriene B4, and platelet-activating factor (108,109). Complement fragments (C5a) can be found in lymph from ischemic myocardium (110), are temporally associated with reperfusion (111), and correlate with increased neutrophil accumulation in reperfused myocardium. Furthermore, C5a directly stimulates neutrophil -O2 production and enhances adherence to coronary artery endothelium (112), whereas C3 activates monocyte adherence (113). The chemoattractant and chemotactic properties of C5a cause progressive generalized vascular leukosequestration (114) during CPB. The interaction between blood and extracorporeal surfaces also stimulates deposition of C3b (115), which in turn amplifies the complement alternative pathway to release C3a and C5a, and subsequently form the membrane attack complex (C5b 9) (115). Accordingly, plasma levels of the anaphylatoxin C5a increase in patients undergoing CPB (115,116), which activates neutrophils to generate -O2 and promotes adhesion to endothelial cells, stimulates degranulation of mast cells, stimulates vascular smooth-muscle contraction, and increases vascular permeability. Complement activation constitutes an early-phase response to CPB, which then progresses
to a second, slower response phase in which de novo synthesis of proteins (E-selectin, ICAM, IL-8) and expression of adhesion molecules on cell surfaces occurs. The magnitude of the complement response has been linked to the duration of CPB and the type of circuitry used (116). The combined stimuli of myocardial ischemia-reperfusion and CPB may synergize to exacerbate postischemic neutrophil-mediated inflammatory responses in patients undergoing CPB for myocardial revascularization.
to a second, slower response phase in which de novo synthesis of proteins (E-selectin, ICAM, IL-8) and expression of adhesion molecules on cell surfaces occurs. The magnitude of the complement response has been linked to the duration of CPB and the type of circuitry used (116). The combined stimuli of myocardial ischemia-reperfusion and CPB may synergize to exacerbate postischemic neutrophil-mediated inflammatory responses in patients undergoing CPB for myocardial revascularization.
Actions of Toll-like Receptors
Recent evidence has shown that activation of TLRs is involved in the pathogenesis of reperfusion injury (117,118). TLRs are transmembrane receptors expressed on endothelial cells, cardiomyocytes, platelets, monocytes, and neutrophils. Cardiomyocytes participate in the inflammatory and innate immune response to ischemia-reperfusion by releasing cytokines (TNFα, IL-1β, IL-6, IL-8, IFN-γ), chemokines, and by expressing cell surface adhesion molecules. TLRs are pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) such as LPS or viral RNA on foreign pathogens, and sense endogenous danger molecules and signals from injured cells. TLRs also recognize and are activated by endogenous ligands such as danger-associated molecular patterns (DAMPS) (e.g., heat shock proteins, HSP70) induced in cardiac tissue during stress (119) and cardiac surgery (120,121). Inducible HSP70 has been found in plasma from CABG patients immediately after surgery (122). ROS may also stimulate TLRs. Ten to eleven human TLRs have been identified; (123,124) TLR receptor-1, -2, -4, -5, and -6 are located on the cell surface, with TLR-2 and TLR-4 being the most involved in ischemia-reperfusion of the group of TLRs; TLR-2 works with other TLRs in co-receptor cooperativity to recognize diacyl- and triacyl-lipoproteins, while TLR4 recognizes LPS from gram-negative bacteria and DAMPs such as HSP70. Neutrophils constitutively express all TRLs except TLR3.
The stimulation of TLRs during ischemia-reperfusion and cardiac surgery can lead to cell injury. Dybdahl et al. (122) reported the release of HSP70 into the circulation of CABG patients and a concomitant increase in monocyte TLR-2 and TLR-4 one day after surgery. Human monocytes exposed to HSP70 released IL-6 and TNFα. Stimulation of TLRs by interaction with DAMPs during ischemia-reperfusion is transduced by numerous molecular signaling pathways, which ultimately results in the activation and translocation of NF-κB to the nucleus, where it stimulates gene expression and release of cytokines and innate inflammatory factors. ROS generated during reperfusion also stimulate TLR-dependent (most likely TLR-4) generation of NF-κB through the PI3-K/Akt pathway, which suggests cross talk between the TLR and reperfusion injury survival kinase (RISK) pathways. Inhibitors of TLRs, intermediary pathways, or NF-κB will attenuate ischemia-reperfusion injury.
The Mitochondrial Permeability Transition Pore as a Determinant of Cell Death
Mitochondria are best known for being the power house of the cell, where ATP is generated through oxidative phosphorylation. However, the mitochondria have been shown to function as a “molecular switch” that governs cardiomyocyte viability; indeed it is a critical fulcrum point in the transition from reversible to irreversible injury. Mitochondria can direct the cell to pursue either an apoptotic or a necrotic pathway to cell death. The key event of this molecular life/death switch is opening of the mPTP. This pore is a transmitochondrial membrane voltage-dependent and Ca2+-dependent high-conductance channel located in the inner mitochondrial membrane that permits water and solutes with a mass up to 1.5 kDa to enter the mitochondrion. Under normal physiologic conditions, the mPTP is in a closed state. It opens in response to high levels of intracellular ROS, Ca2+ and inorganic phosphate when mitochondria matrix and intracellular pH are in the normal range. The mPTP remains closed during ischemia because intracellular acidosis and the intracellular levels of Mg2+ and ADP counteract the opening triggered by ROS and Ca2+. However, the mPTP opens during the early moments of reperfusion (125) when there is an accumulation of ROS, Ca2+ and inorganic phosphate, and when there is a rapid renormalization of intracellular and mitochondrial matrix pH (126). The opening of the mPTP increases the permeability of mitochondria to water and small solutes, causing swelling of the matrix and collapse of the mitochondrial transmembrane potential that drives oxidative phosphorylation and ATP generation. mPTP opening also stimulates the release of the pro-apoptotic factors cytochrome c and apoptosis-inducing factor (AIF). The switch function comes into play based on the availability of intracellular ATP; if ATP is present then the cell pursues an apoptotic pathway because apoptosis requres ATP. If ATP levels are depleted then the cell pursues a necrotic pathway. With prolonged antecedent ischemia mPTP is irreversible, but milder or short periods of ischemia trigger transient reversible opening (127).
PHYSIOLOGIC CONSEQUENCES OF REPERFUSION INJURY
The multiple etiologies of reperfusion injury following nonsurgical or surgical reperfusion can present as numerous abnormalities in electrophysiology, contractile function, biochemistry, and morphology. Ischemia-reperfusion injury falls into reversible and lethal injuries. Reversible injury includes impaired contractile and relaxation mechanics (e.g., low output syndrome) in the absence of cell death (stunning), edema, and arrhythmias. Lethal injury includes apoptosis and necrosis. Myocardium can transition from reversible to irreversible injury by increasing the duration of ischemia, or by the advancing penumbra of the reperfusion “wavefront.” In any event, reperfusion injury begins within the first few minutes of the onset of reperfusion or even the elimination of ischemic
conditions due to manipulation of oxygen demand. It progresses over approximately 24 hours in larger animal models and, presumably, in humans. The clinical manifestations of ischemic-reperfusion injury can be grouped into the categories listed below.
conditions due to manipulation of oxygen demand. It progresses over approximately 24 hours in larger animal models and, presumably, in humans. The clinical manifestations of ischemic-reperfusion injury can be grouped into the categories listed below.
Reperfusion Dysrhythmias
Benign or more malignant arrhythmias and conduction disturbances may be observed following significant ischemia followed by reperfusion. Varying degrees of loss of automaticity requiring temporary pacing strategies, heart block, and arrhythmias manifested as premature ventricular contractions, ventricular tachycardia, or ventricular fibrillation may be observed. Clearly, with the previous discussion on ionic imbalance, the reader can appreciate the degree to which the movement of Na+, K+ and Ca2+ ions can be deranged in this type of setting. It is not at all surprising that postoperative/postprocedure arrhythmias are a frequent complication of both adult and pediatric cardiac surgery (128,129), acute coronary syndromes and various types of reperfusion therapy, and such can contribute to postoperative morbidity and mortality. The financial burden of anti-arrhythmic therapy is significant. Atrial fibrillation is a consequence of cardiac surgery affecting ˜30% or more of all surgical patients, whose treatment reaches billions of dollars. We will not discuss atrial fibrillation further in this chapter because it is complex and its relation of cardioprotective strategies is not yet clear. Failure of spontaneous resumption of sinus rhythm, ventricular fibrillation, and persistence of dysrhythmias requiring direct-current countershock and anti-arrhythmic therapy are common consequences of inadequate myocardial protection.
A number of mechanisms are involved in the genesis of reperfusion dysrhythmias. First, as with many reperfusion-related events, the incidence and severity of reperfusion dysrhythmias relate to the severity of the preceding ischemia and consequent reperfusion injury. The relationship is characterized by a bell-shaped response curve, in which vulnerability to arrhythmias is maximal after short intervals of normothermic ischemia induce reversible changes, then decreases as irreversible injury and electrical silence develop with more prolonged ischemia. Second, an accumulation of intracellular Ca2+ during ischemia may interfere with normal Ca2+ cycling at the level of the sarcoplasmic reticulum (Ca2+-dependent arrhythmias). Third, oxygen-derived free radicals such as -O2 and •OH, produced as a respiratory burst by mitochondria, endothelium, or neutrophils during reperfusion, can damage membrane lipids and various transport proteins involved in ionic homeostasis, which in turn can precipitate reperfusion-related dysrhythmias (130,131). Current hypotheses merge the Ca2+-related events and the ROS-related events as “interacting triggers” for reperfusion dysrhythmias. Fourth, hyperkalemia is also a contributing factor to postoperative arrhythmias (reviewed in Dobson et al. (27)) as suggested by the correlation found by Ellis and coworkers between potassium concentration in cardioplegia solutions and the incidence of arrhythmias (132) There is an anatomical hierarchy of sensitivity to potassium, with the atria being most sensitive and the sino-atrial node and internodal tracts being less sensitive. Differences in sensitivity to potassium also exist across the ventricular wall at the base and apex of the heart, where repolarization times are less in the basal epicardium versus the endocardium during hyperkalemia. These transmural differences in repolarization times may lead to arrhythmias on rewarming and reanimation.
Postischemic Systolic and Diastolic Dysfunction
Reperfusion with unmodified blood following short periods of normothermic regional or global ischemia produces a severe degree of contractile dysfunction (Fig. 9.2A, B). While short periods of ischemia followed by unmodified reperfusion may be benign in regard to contractile function, it may be injurious to other targets (endothelium, mitochondria). Readily reversible (within hours or days) postischemic contractile dysfunction can occur in the absence of obvious morphologic injury and may therefore be “stunned,” whereas longer periods of ischemia are associated with both morphologic injury (necrosis, apoptosis) and persistent contractile dysfunction. Previously, postischemic contractile dysfunction was thought to be attributable to a depletion of high-energy phosphate supply (i.e., ATP). However, numerous studies have failed to show a direct correlation between postischemic myocardial ATP levels and contractile function. The ATP turnover rate, roughly approximated by myocardial oxygen consumption, may be more important as a barometer of oxidative rate than the static tissue level of ATP. Recently, postischemic contractile dysfunction has been more related to impairment of Ca2+ kinetics in excitation-contraction coupling and in the sarcoplasmic reticulum (133). In addition to these pathologic origins of postischemic systolic contractile dysfunction, iatrogenic dysfunction related to CPB per se may occur as a result of systemic hemodilution (reduced oxygen delivery) and systemic hyperkalemia, or as a result of systemic hypocalcemia caused by Ca2+ chelating agents in cardioplegia solutions.
Myocardial diastolic characteristics (compliance or chamber stiffness, rate of relaxation) are highly sensitive to ischemia and reperfusion. In the ischemic myocardial segment, there is a rightward shift in the position of the end-diastolic pressure-segment length relation, with little change in the shape or curvature of the end-diastolic pressure-segment length relation, which is consistent with the concept of regional myocardial “creep” (Fig. 9.2A. Other reports show little or no change in segmental stiffness after up to 1 hour of coronary occlusion (134,135). In contrast to ischemia, reperfusion is associated with immediate loss of myocardial compliance (135), characterized by a leftward shift and increase in slope of the
exponential pressure-segment length relationship (indicating a degree of local “contracture”). A similar pattern is observed with global ischemia. In several studies in which hearts were sensitized to reperfusion injury by 30 minutes of normothermic ischemia preceding 1 hour of hypothermic (4°C) multidose blood cardioplegia, a rightward shift in the end-diastolic pressure-dimension relations (dilation) and increased chamber stiffness were noted after blood reperfusion (136,137) (Fig. 9.2B). Acute increases in postischemic chamber stiffness are caused by myocardial edema and abnormal Ca2+ handling in the myocardium. A decrease in diastolic relaxation impairs diastolic ventricular filling, which reduces stroke volume independent of any postischemic or postcardioplegia abnormalities in inotropic state or contractility.
exponential pressure-segment length relationship (indicating a degree of local “contracture”). A similar pattern is observed with global ischemia. In several studies in which hearts were sensitized to reperfusion injury by 30 minutes of normothermic ischemia preceding 1 hour of hypothermic (4°C) multidose blood cardioplegia, a rightward shift in the end-diastolic pressure-dimension relations (dilation) and increased chamber stiffness were noted after blood reperfusion (136,137) (Fig. 9.2B). Acute increases in postischemic chamber stiffness are caused by myocardial edema and abnormal Ca2+ handling in the myocardium. A decrease in diastolic relaxation impairs diastolic ventricular filling, which reduces stroke volume independent of any postischemic or postcardioplegia abnormalities in inotropic state or contractility.
Myocardial Necrosis
Necrosis is “accidental” injury to the cell in which the cell swells and the sarcolemma bursts. The process of necrosis does not require energy. In histologic terms, local hypercontracture occurs which is consistent with contraction bands. Intracellular contents are released into the extracellular space and subsequently into the circulation. Large molecules such as creatine kinase (MB isoenzyme) and cTnT or cTnI released into the circulation have been used as biomarkers of cell death by necrosis. In cardiac surgery, infarction is largely localized to the previously ischemic and revascularized (or non-revascularizable) segments of myocardium. Global necrosis related to contracture may be observed as stone heart. Release of cardiac enzymes during surgery above that observed preoperatively suggests that myocardium suffers necrosis (diffuse global or discrete regional infarction) despite restoration of blood flow, and suggests further that myocardial protection is not optimal. The ARTS (Arterial Revascularization Therapies Study) trial linked the increased CK release to clinical mortality (138). The association of increased plasma CK and other markers with clinical outcomes and mortality has been confirmed by other studies (139,140,141,142,143). A meta-analysis showed that increased release of plasma biomarkers was associated with 1-year and >3-year risk of mortality (144). CK-MB, cTnI, or cTnT have often been used in clinical studies as end points to discriminate benefits my myocardial protection strategies.
Apoptosis
In contrast to necrosis, apoptosis (Greek for “dropping off of leaves”) is a form of cell death that is highly regulated and energy-requiring (145). Cells targeted for apoptosis undergo DNA fragmentation in specific areas by endonucleases which cause condensation of nuclear material rather than the disintegration seen in necrosis; the cell begins to break apart in discreet membrane-enclosed vesicles (apoptotic bodies) which results in shrinkage of the cell, with ultimate phagocytosis by macrophages without disturbing nontargeted cells next door. The morphologic changes of apoptosis can be observed as after less than 2 hours after exposure to the stimulus, and can last for 12 to 24 hours. The average adult human will lose between 50 and 70 billion cells each day by apoptosis. Apoptosis is responsible for cell dropout during development such as resorption of a tadpole tail, or removal of webbing between the phalanges to define the fingers during fetal development. Likewise, mature cells that are damaged or dysfunctional are removed by apoptosis. Apoptosis is known to be a major cause of progressive heart failure (146), local infarction, cardiomyopathy, myocarditis, and ischemia-reperfusion of the myocardium (147). Apoptosis has been reported in patients undergoing cardiac surgery (148,149,150,151,152,153). It has been linked to cardiac dysfunction and stunning after cardiac surgery (see an excellent review by Anselmi et al. (154)). In 2000, Aebert et al. (149) reported that plasma from patients undergoing CPB caused apoptosis in cultured endothelial cells, suggesting that the substances that induce apoptosis are carried in the blood likely as cytokines, as well as originating locally.
Apoptosis follows either extrinsic or intrinsic pathways. In the extrinsic pathway, apoptosis is stimulated by FAS (first apoptosis signal) and inflammatory mediators interacting with membrane receptors. TNFα is a major stimulator of the extrinsic pathway by interaction with TNFα receptor type 1 (TNF-R1) or 2 (TNF-R2). The FAS-FAS ligand pathway is engaged when FAS ligand (a member of the TNF family) binds to its cognate FAS receptor to ultimately stimulate downstream apoptosis-activating proteins (caspases). Some of these factors that trigger the extrinsic pathway such as IL-6, IL-8, TNFα (155), and FAS are released during CPB and/or chemical cardioplegia. The intrinsic pathway is stimulated by oxidants, hypoxia and ischemia, and reperfusion. Evidence suggests that apoptosis is initiated by ischemia, but executed at reperfusion (156,157). Ischemia-reperfusion stimulated apoptosis processes are regulated by pro (bcl2) and anti (bax) apoptotic proteins; this pro- and anti-regulatory scheme provides a degree of prevention and reversibility to the demise of the cell, unlike necrosis. However, the cleavage of downstream cysteine-dependent protease enzymes, notably caspase 8, 9, and 3, executes the apoptotic cell death program. The extrinsic and intrinsic pathways ultimately converge on the mitochondria, which undergo either swelling or opening of the mPTP, and release pro-apoptotic cytochrome c and AIF. The AIF pathway is independent of the caspase activation cascade.
Consistent with the conditions that initiate apoptosis, inhibitors of oxidant generation or activity and, more recently, inference regarding avoidance of over-oxygenation, can mitigate apoptosis in surgical experimental models (158,159,160,161). ATP-sensitive potassium (KATP) channel openers have also been found to reduce apoptosis in surgical cardioplegia experimental models (162,163). In addition, endonuclease inhibitors such as aurintricarboxylic acid (ATA) attenuate the development of myocardial apoptosis after ischemia-reperfusion.
However, these and other anti-apoptotic therapies have yet to be tested in human cardiac surgery.
However, these and other anti-apoptotic therapies have yet to be tested in human cardiac surgery.
A significant postischemic, postreperfusion increment of injury is known to lead to unfavorable remodeling of the left ventricle. The contribution of such injury to the development of ischemic cardiomyopathy after myocardial infarction is not disputed and therapies to avoid this type of injury are discussed further below.
Endothelial Dysfunction
Effects of Ischemia-Reperfusion on NO• Homeostasis and Endothelial Dysfunction
The general assumption has been that the consequences of ischemia-reperfusion injury affect primarily the cardiac myocyte. However, abundant data indicate that ischemia and reperfusion affect other cell types in the heart, notably the coronary vascular endothelium. For example, the vascular endothelium of both arteries (91,164) and veins (165) sustains significant injury during ischemia and reperfusion, and reduces both basal and stimulated NO• release (164,166). The endothelium may be the primary site of complement activation and neutrophil activity after ischemia and reperfusion. This dysfunction may persist beyond the immediate acute phase of reperfusion for days and weeks (167) following ischemia-reperfusion. Endothelial dysfunction plays a critical role in the pathogenesis of myocardial reperfusion injury and infarction (168,169).
Data presented by Malinski and Taha (170) suggest that there is an initial burst of NO• release during early ischemia that consumes local substrate L-arginine and the cofactor BH4; depletion of substrate and cofactor is associated with the uncoupling of eNOS and NO• generation in favor of –O2 generation by transfer of the electron to molecular oxygen rather than to nitrogen (171). This -O2 may quench NO• to form ONOO- at reperfusion, thereby further depleting local NO• and creating NO• dyshomeostasis. It is possible that ischemia associated with unprotected global ischemia or intermittent cardioplegia is associated with a similar uncoupling of eNOS and generation of –O2.
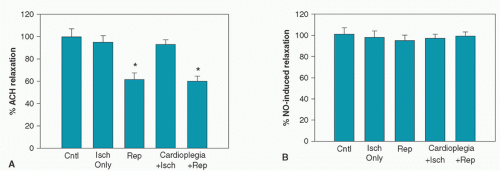 FIGURE 9.5. Relaxation response of epicardial coronary arteries to agonist stimulators of nitric oxide synthase preconstricted with U46619. A: Maximal relation responses to acetylcholine (ACH), and endothelium-dependent, receptor-dependent agonist. B: Maximal relaxation responses to the direct (endothelium-independent) smooth muscle relaxing agent acidified NaNO2. Cntl, normal control artery; Isch Only, after 45 minutes of global normothermic ischemia (cross-clamping); Rep, ischemia was followed by 1 hour of unmodified blood reperfusion; Cardioplegia + Isch, 45 minutes of global normothermic ischemia followed by 1 hour of intermittent (q 20 minutes) 4°C hyperkalemic blood cardioplegia without reperfusion (declamping); Cardioplegia + Rep, cardioplegia as described previously followed by 1 hour of reperfusion (declamping). *p < 0.05 versus unstarred groups. (From Nakanishi K, Zhao Z-Q, Vinten-Johansen J, et al. Coronary artery endothelial dysfunction after ischemia, blood cardioplegia, and reperfusion. Ann Thorac Surg 1994;58:191-199, with permission.) |
Since NO• has pleiotropic effects in the cardiovascular system, impaired release is expected to have multiple consequences. Impaired release of NO• is expressed physiologically as an increase in the adherence of neutrophils to the surface of the endothelium and impaired vasodilator responses to endothelium-specific stimulators of NO• synthase, such as acetylcholine. Decreases in in vitro NO•, measured directly following ischemia-reperfusion, parallel the increased adherence of neutrophils to coronary artery endothelium and blunted endothelium-dependent vasodilator responses to acetylcholine and bradykinin (86,172). Figure 9.5 shows coronary artery segment function following 45 minutes of global normothermic ischemia with and without blood reperfusion (release of cross-clamp). Endothelial function, assayed as maximal vasorelaxation responses to the endothelium-dependent, receptor-dependent agonist acetylcholine, was not significantly reduced in hearts subjected to ischemia without reperfusion, but were significantly reduced in reperfused hearts in comparison with controls (164). These data suggest that endothelial damage occurs during reperfusion rather than during shortterm global ischemia. At least a portion of this postcardioplegia endothelial dysfunction may be induced by CPB per se. Zanaboni et al. (173) reported that pulmonary endothelial function is impaired for 3 to 4 days after CPB. Inflammatory
mediators (174) and gaseous microemboli (175) may be involved in this endothelial injury by CPB.
mediators (174) and gaseous microemboli (175) may be involved in this endothelial injury by CPB.
Myocardial Edema
Myocardial ischemia and reperfusion are associated with abnormalities in the regulation of intracellular and interstitial fluid balance. Specifically, certain aspects of these mechanisms, including the Starling forces governing tissue fluid movement, lymphatic drainage, and cell membrane function, are altered to favor water retention in the cell or extravasation into the interstitial space. Although the pathophysiology involved in producing myocardial edema during ischemia-reperfusion injury has not been completely elucidated, Garcia-Dorado et al. (176) and others (177) have observed an increase in myocardial water content following coronary occlusion-reperfusion in animal models. Although some cell swelling may occur during ischemia, major intracellular and interstitial edema formation seems to be a reperfusion phenomenon (176,178,179). Potential mechanisms of myocardial edema during ischemia-reperfusion include the following: (1) increased intracellular osmotic pressure secondary to an accumulation of metabolic end-products of anaerobic glycolysis, lipolysis, and ATP hydrolysis; (2) interference during ischemia with the maintenance of electrical potentials across cell membranes, in which an intracellular accumulation of Na+ and Cl– (ionic dyshomeostasis) leads to a movement of water into the intracellular space; (3) increases in both microvascular permeability and osmotic pressure within the interstitial space. Water may not only be passively regulated but may be regulated by several membrane pores called aquaporins. Select aquaporins (types -1, -3, and -7) are expressed in human hearts (180). Aquaporins facilitate the movement of
water between spaces along osmotic gradients, and function in reabsorption of water by the kidneys. Recently, focus has been aimed at the role of aquaporins in volume regulation in endothelial cells and myocytes, as well as between vascular, interstitial and lymphatic spaces during ischemia and reperfusion (180,181,182). However, aquaporins in endothelial cells and cardiomyocytes may have a role in edema of those tissues. The inhibition or experimental (gene) deletion of aquaporins reduces edema, suggesting that these membrane pores may be a future target of therapy in surgical and nonsurgical ischemia-reperfusion injury.
Edema may increase microvascular resistance to a point of impeding blood flow (no-reflow phenomenon, discussed below) and increase diffusion distance to myofibrils, leading to inadequate oxygen delivery at the tissue level. Edema can arise with the use of cardioplegic solutions, especially in ischemic myocardium, because of (1) high delivery pressures, particularly in severely damaged myocardium; (2) hemodilution and hypo-osmolarity from crystalloid primes or crystalloid cardioplegia solutions; (3) physiologic changes in ionic pump systems (i.e., Na+-K+ ATPase) or Donnan equilibrium for chloride ions induced by hypothermia; and (4) decrease in lymphatic drainage during arrest. Although normal myocardium tolerates relatively high infusion pressures, myocardium within (135,183) and surrounding (184) ischemic segments is vulnerable to edema induced by high delivery pressure. The extent to which reperfusion with unmodified blood following CPB induces myocardial edema depends primarily on the duration and severity of the previous ischemic episode (176). On reperfusion, the normo-osmotic blood quickly displaces the hyperosmotic intravascular fluid, creating an osmotic gradient between the intravascular and extravascular spaces that results in myocardial edema (179). Myocardial edema following ischemia and reperfusion is also partly caused by an influx of Na+ ions into the cell, accompanied by interstitial water. Correspondingly, the Na+-H+ exchanger plays a major role in producing a net influx of Na+ into the cell, which leads to intramyocardial edema (178,185). Inhibition of this Na+-H+ exchange system has been shown to decrease postischemic myocardial edema (61).
Cardioplegia may contribute to edema formation in ischemically injured hearts. The composition of cardioplegic solutions (onconicity, hemodilution) and the conditions of delivery (hypothermia, high delivery pressure) are known to exaggerate the development of edema resulting from ischemia or systemic inflammatory responses.
Microvascular Injury and the “No-Reflow” Response
One of the physiologic consequences of reperfusion injury is impaired microvascular blood flow. Tissue blood flow defects after ischemia were first reported by Krug et al. (186) in 1966, and the name no-reflow was later applied by Ames and colleagues (187) in 1968. In 1974, Kloner et al. (188) described the “no-reflow” phenomenon as a derangement in postischemic blood flow to the myocardium at risk despite resolution of the coronary obstruction. The etiologies of the “no-reflow” or “low-reflow” phenomenon include (1) postischemic tissue edema and interstitial hemorrhage compressing the vascular space, (2) active vasoconstriction from loss of endothelium-derived vasodilators and release of neutrophil-derived vasoconstrictors, (3) capillary plugging by neutrophils adhering to a dysfunctional endothelium, and (4) air emboli and/or intravascular debris (detached endothelial cells, clot, atherosclerotic plaque) obstructing blood flow. Microvascular injury and postischemic defects in myocardial blood flow may be a reperfusion phenomenon because (1) the absence of a significant perfusion pressure during ischemia prevents the migration of fluid out of the extravascular space, so that edematous fluid fails to accumulate significantly, (2) endothelial damage with adherence and trapping of neutrophils and loss of NO-induced regulation of vascular resistance occurs during the early moments of reperfusion (91), and (3) the boundaries of no-reflow zone widen as reperfusion continues (189) consistent with the “wavefront” of ischemia-reperfusion injury.
In surgery, perfusion pressure is important not only at the release of cross-clamp, but during delivery of cardioplegia. Delivery pressures of 150 to 200 mm Hg measured proximal to the cardioplegia delivery cannula are sometimes experienced; aortic root pressure will be somewhat less than this because of the relatively high resistance at the catheter tip. However, it would be important to balance the vulnerability of the myocardium to pressure-induced damage with adequate pressures necessary to ensure delivery of cardioplegia solution.
STRATEGIES TO PROTECT THE HEART DURING CARDIAC SURGERY
Role of Cardioplegia in Reducing Surgical Ischemic-Reperfusion Injury
Cardioplegia offers selective perfusion to the heart and the administration of agents in higher concentrations that may otherwise not be well tolerated by other organs or systemically without complications (e.g., toxicity, hypotension). During selective cardiac delivery, drugs at cardioprotective or anti-inflammatory concentrations may be diluted below effective or active concentrations after admixture with systemic blood. This is certainly the case with the most basic component of cardioplegia, K+, which is often administered at greater than five times the normal blood concentration. It is important to understand that cardioplegia exerts cardioprotection during both ischemia and reperfusion because of the intimate link between ischemic injury and reperfusion injury.
Cardioplegia exerts cardioprotection by modifying the conditions of (re)perfusion and the composition of the perfusate (cardioplegia solution) or reperfusate (terminal cardioplegia, hot shot) if given during the early moments of reperfusion to reanimate the heart. In its simplest formulations, cardioplegia avoids ischemic injury primarily by reducing oxygen demands to less than 10% of that of the working heart by (1) producing immediate asystole, (2) rapidly initiating hypothermia, (3) providing intermittent reoxygenation (multidose regimens), and (4) improving anaerobic metabolism (190). Cardioplegia also avoids reperfusion injury by targeting specifically the pathophysiologic mechanisms and mediators involved in ischemia-reperfusion injury. Beneficial modifications to the conditions of cardioplegia infusion apply to infusion pressure and volume, duration of infusion, and temperature of the solution. Modifications to the composition of cardioplegia during delivery or at reperfusion include addition of metabolic substrates, osmotic agents, buffers, and cardioprotective pharmacologic agents, as well as whether K+ is used as the arresting agent or some nondepolarizing arresting agent (see below). Cardioplegia can be viewed as a vector for pharmacologic therapies to the heart with agents that target specific aspects of either ischemic or reperfusion injury.
The inclusion of any pharmacologic agent in a cardioplegic solution should be based on scientific principles tested in the laboratory and supported by literature. There should be a defined target(s) toward which the agent or additive is aimed. The timing of pharmaceutical delivery is also very important. “What comes first must be treated first” (191) is a mantra worth using as a guide to deliver therapies at the appropriate time relevant to the pathophysiologic events of ischemia and/or reperfusion injuries. To administer a pharmacologic agent either before or after the occurrence of its targeted event is a missed opportunity. If the therapy is designed to exert actions on mechanisms operative during ischemia, administration of the agent at reperfusion or after declamping may be ineffective. On the other hand, if events are occurring during early reperfusion, administration during later reperfusion will be ineffective. In addition, the physiologic environment must be considered, such as hypothermia versus normothermia, or blood versus crystalloid. Although hypothermia has numerous advantages, as discussed below, it also reduces membrane fluidity and potentially attenuates the ligand-receptor interactions on which the effects of many drugs depend. Therefore, the effectiveness of certain cardioprotective drugs can be attenuated when hypothermic strategies are used. Finally, adequate delivery is extremely important, particularly to areas with occlusive coronary disease, because the cardioplegic solution cannot exert the effect for which it is designed if it is not adequately delivered to the target area.
Phases of Myocardial Protection
There are four phases of myocardial protection in cardiac surgery: pretreatment, induction, maintenance, and reanimation, consistent with the time points of injury shown in shown schematically in Figure 9.1.
Pretreatment therapy includes ischemic and pharmacologic preconditioning (192). Pharmacologic pretreatment or preconditioning would include volatile anesthetic agents or propofol which exert protection at the cellular and mitochondrial levels (193).
The induction phase is intended to not only arrest the heart and reduce myocardial temperature, but is also an opportunity to resuscitate the heart that is energy-depleted or failing (194). Warm induction is an example of a therapeutic role of the induction phase. This is discussed further below.
The maintenance of arrest is an opportunity to reoxygenate and restore nutrients to the myocardium, wash out metabolic byproducts, and restore ionic balance to the myocardium. Maintenance can be achieved by intermittent infusions of solution, for example, every 20 to 30 minutes, or continuous infusion, usually in the retrograde mode.
The reperfusion and reanimation period at the time of declamping is very important to the overall conduct of myocardial protection (27), but is a 5- to 10-minute interval that is sometimes underappreciated. It is an opportunity to remove or lessen the thermal gradient between the
cold heart and the warm systemic blood, establish ionic homeostasis, increase the aerobic metabolic capacity after suppression by hypothermia, repolarize the heart if hyperkalemic depolarizing arrest is used, and to deliver drugs that work specifically at reperfusion. The avoidance of arrhythmias and the use of direct current countershocks may well depend on how the reperfusion and reanimation period is conducted. Controlled perfusion through the cardioplegia cannula with the clamp still on has been used to provide warm blood and cardioprotective drugs to the heart. It is not necessary to maintain arrest during this period as has been practiced in the past since the heart is being perfused continuously.
What End Points Should Cardioprotective Strategies Target?
Surgical mortality has been driven to historically low levels, especially in low-risk isolated CABG. Although mortality rates are slightly greater in high-risk patients with multiple procedures, longer cross-clamp times, poor or failing preoperative ventricular performance, or advanced age (195), even highrisk cohorts face mortality risk much lower than ever before. In today’s healthcare environment, however, longer-term measures of ventricular function, subsequent admissions and events as well as nonmortality complications will be scrutinized. These represent new and ongoing challenges to the outcomes of surgery, and allude to costs and resource utilization. Postoperative atrial fibrillation continues to be a nuisance with an incidence of approximately 20% to 40% with a cost of over $1.5 billion in the US alone (196). Perioperative stroke remains a rare but devastating complication of cardiac surgery, but is not directly related to cardiac protection. Hence, while in-hospital and 30-day mortality is still an important metric, others are also being viewed as critical to the yardstick of appropriate surgical outcomes. Cardioprotective efforts during surgery should focus on reducing the ischemia-reperfusion injury, insuring improvement rather than diminishment in postischemic cardiac function, and reducing the contribution to the heart failure epidemic especially in patients operated on during evolving myocardial infarction. Relative to mortality alone, postischemic heart failure represents the larger part of the iceberg that is under the surface (27).
Formulations of Cardioplegia
Approximately 98% of cardiac surgeons use some form of chemical cardioplegia (197), with 72% of surgeons in the United States using blood cardioplegia and 28% using crystalloid cardioplegia. There are no formal federal guidelines for cardioplegia solution composition or method of use historically. In fact, in the past, the FDA has classified cardioplegia solutions as a “medical device”; however, more rigorous testing in vitro and in in vivo models incorporating CPB is being requested by the FDA. However, there are hundreds of solutions and methods of use based on mentor teaching, personal preference, and either experimental or clinical experience. Based on a limited survey in Missouri (198), there is little uniformity in formulation of cardioplegia solutions, with many being formulated by hospital pharmacies despite availability of FDA-approved compounding pharmacies.
Cardioplegia vehicles are categorized as crystalloid or blood, and the crystalloid formulations are further subdivided into intracellular or extracellular ionic compositions.
Crystalloid Cardioplegia
Celsior
The introduction of Celsior by Philippe Menasché in 1994 used the isolated rat heart that was arrested, placed in cold storage for 5 hours, and reanimated to obtain recovery of left ventricular developed pressure, LV dP/dt, and compliance post-storage. The data from this isolated-perfused preparation was recapitulated in other small animal preparations (199), in large animal in situ isolated-perfused studies (200), in transplant models (201,202), and was finally translated in human clinical trials (203,204,205,206). Currently, Celsior is used primarily for preservation during cold storage.
Plegisol
Plegisol is the commercial name for St. Thomas’ II solution, formulated and tested by David Hearse (18,21,207). It was clinically validated in 1989 (208); it is sold commercially in the United States by Hospira. This crystalloid solution is used for clinical cardioplegia internationally, specifically in Britain, variably in other European countries, and in about 15% of cases in the US.
Del Nido Solution
The del Nido solution, developed by Dr. Pedro del Nido, was patented in 1995 (USPTO # 5,407,793). It was developed as a cardioplegia solution for protection of immature hearts (209), but its use has been extended to all hearts in Boston Children’s Hospital (210,211) and other institutions (212,213). The base of the solution is Plasma-Lyte A (Baxter Healthcare) which has electrolyte composition similar to that of extracellular fluid. The salient compositional features of the del Nido Solution are low Ca2+ content (provided by the 20% of volume blood), hyperkalemia (24 mEq/L); mannitol for oxygen radical scavenging and osmotic properties, sodium bicarbonate for buffering of acidosis during arrest; and lidocaine to stabilize membrane potential and reduce arrhythmias. Its use as a cardioplegia solution in adult patients is currently under investigation, but contemporary studies involve small numbers of patients, are retrospective in design, and show only safety and noninferiority to other solutions based on lack of significant differences between del Nido solution and other solutions (214,215,216).
Custodiol
Custodiol is the former Bretschneider solution, also called HTK (histidine, tryptophan, ketoglutarate) solution. It is an
intracellular composition crystalloid solution originally developed as a cardioplegia solution in Germany (217), but now used for perfusing and flushing of organs from the donor, preservation of organs for transplant (218), and for cardioplegia in cardiac surgery. Custodiol was successfully translated into large animal simulations of cardioplegia (219) and transplantation, which predicted success in subsequent clinical trials and reports (220,221,222,223).
intracellular composition crystalloid solution originally developed as a cardioplegia solution in Germany (217), but now used for perfusing and flushing of organs from the donor, preservation of organs for transplant (218), and for cardioplegia in cardiac surgery. Custodiol was successfully translated into large animal simulations of cardioplegia (219) and transplantation, which predicted success in subsequent clinical trials and reports (220,221,222,223).
Blood Cardioplegia
Hemodiluted Blood Cardioplegia
Blood is viewed by some as an “ideal” vehicle to carry cardioplegia components because blood has (1) an increased carrying capacity to provide greater oxygen delivery to maintain oxygenation and repay oxygen debt; (2) an innate buffering capacity from histidine moieties; (3) “natural” osmotic properties that exceed that of crystalloid solutions; (4) blood-borne nutrients such as glucose and free fatty acids; and (5) endogenous oxygen radical scavengers such as glutathione and glutathione peroxidase, and catalase. On average, O2 consumption of the normally working heart is ˜8 mL O2/min/100 g, or 28 mL/min for a 350 g heart. In contrast, in a crystalloid solution, 700 mL/min of cold (10°C) solution/min would be required to deliver this much oxygen based on an O2 carrying capacity for crystalloid solution of 4 mL O2/100 mL. Although the argument has been made that O2 bound by hemoglobin is not available at cold temperatures because hypothermia shifts the hemoglobin dissociation curve to the left, experiments have shown that oxygen is indeed released at cold temperatures during delivery of cardioplegia (137), ostensibly facilitated by a low tissue PO2, and the effects of pH and 2,3-DPG on oxygen dissociation from hemoglobin. The first blood cardioplegia solutions were hemodiluted with crystalloid-dissolved agents in a 4-parts blood-to-1-part crystalloid ratio (224). Newer delivery techniques have incorporated methods to continuously blend blood from the CPB reservoir with the contents of the crystalloid bag in any number of given ratios or variable ratios (e.g., the Quest MPS unit). With the recent focus on limiting the crystalloid load, higher ratios have been used clinically.
Blood cardioplegia maintains an aerobic arrest which is important to avoiding ischemia and reperfusion injuries. Accordingly, we found that oxygen was a critical factor in the benefits of cold-blood cardioplegia (137). However, blood does contain inflammatory cells (i.e., neutrophils) and pro-inflammatory cytokines. Whether these potential negative effects are translated to postcardioplegia injury depends in large part on avoidance of ischemia, which sensitizes the myocardium to other aspects of reperfusion- and inflammatory-based injuries.
All-Blood Cardioplegia
Menasché and colleagues have coined the term “miniplegia,” now referred to as “microplegia,” to describe a technique of administering a small quantity of a prepared arresting solution via a pump-driven syringe to otherwise unmodified blood originating from the CPB circuit (225,226,227). The volume and delivery rate of the concentrated potassium (and magnesium) solution is incrementally decreased to the lowest level possible to maintain quiescence. The key to this type of myocardial protection is the nearly continuous delivery of the blood solution to the myocardium at a tepid temperature. The continuous administration obviates the need for added buffers such as tromethamine (THAM) and citrate-phosphate dextrose because aerobic metabolism is maintained throughout the duration of arrest, resulting in sufficiently high production of energy to drive the ionic pumps responsible for Na+, Cl– and Ca2+ homeostasis, and avoid intracellular acidosis. The reduction in crystalloid “diluants” avoids progressive hemodilution.
The potential benefit of microplegia solutions are numerous and include (1) increased oxygen supply and increased presence of endogenous antioxidants and detoxification factors lost with hemodilution; (2) improved control of blood volume to reduce edema resulting from hypo-osmolality, bleeding complications, and potassium overdose; (3) increased convenience and practicality; and (4) better cost effectiveness. Reports indicate good clinical satisfaction with this technique (228). This philosophy of “all-blood cardioplegia,” however, does not inherently embrace the application of additives addressing mediators of ischemia-reperfusion injury that may be of clinical benefit. In addition, this “microplegia” philosophy challenges the need for other strategies of myocardial protection developed during the past two decades, including buffering of intracellular acidosis, calcium management, and use of hyperosmolar conditions. However, perhaps a resolution to this conundrum is avoidance of ischemia altogether, which prevents injury from occurring in the first place. These issues bear further examination, as they relate to newer strategies of myocardial protection.
Major Strategies of Protection with Cardioplegia
The major characteristics of cardioplegia solutions in regard to the phases of protection, actions of constituents, and methods of achieving the actions are summarized in Table 9.1.
Depolarized Arrest Using Hyperkalemia
Exogenous potassium causes a concentration-dependent depolarization of the cell transmembrane potential. For example, at 10 mM (10 mEq/L) the transmembrane potential is depolarized from its normal -85 mV to approximately -65 mV, rendering the myocardium unexcitable. At this transmembrane potential the voltage-dependent fast Na+-channels are blocked, and there theoretically is no phase 0 of the action potential, and hence no generation of the action potential; this is a state of “depolarized” arrest. Potassium concentrations in cardioplegic solutions ranging from 12 to 30 mEq/L are typically used to achieve cardiac standstill within 1 to 2 minutes under hypothermic conditions, although more moderate levels of potassium have been reported (229). Higher potassium
concentrations are used to arrest the heart with normothermic induction techniques (230) or to maintain arrest during a warm reperfusate cardioplegia (i.e., “hot shot”) delivered just before aortic declamping. Potassium concentrations above 40 to 50 mEq/L have been avoided because of the potential for tissue and coronary vascular endothelial damage, although this point remains unsettled. Washout of hyperkalemic cardioplegic solutions by noncoronary collateral flow, or an increase in myocardial temperature permits resumption of both electrical and mechanical activity, which may escape detection and can be counteracted by intermittent (every 20-30 minutes) replenishment of cardioplegic solutions. Such a multidose approach to infusion of cardioplegia solutions maintains or re-establishes electromechanical silence and pays additional dividends of restoring hypothermia, providing the ischemic myocardium with oxygen (aerobic arrest) and nutrients, and washing out metabolites of anaerobiosis.
concentrations are used to arrest the heart with normothermic induction techniques (230) or to maintain arrest during a warm reperfusate cardioplegia (i.e., “hot shot”) delivered just before aortic declamping. Potassium concentrations above 40 to 50 mEq/L have been avoided because of the potential for tissue and coronary vascular endothelial damage, although this point remains unsettled. Washout of hyperkalemic cardioplegic solutions by noncoronary collateral flow, or an increase in myocardial temperature permits resumption of both electrical and mechanical activity, which may escape detection and can be counteracted by intermittent (every 20-30 minutes) replenishment of cardioplegic solutions. Such a multidose approach to infusion of cardioplegia solutions maintains or re-establishes electromechanical silence and pays additional dividends of restoring hypothermia, providing the ischemic myocardium with oxygen (aerobic arrest) and nutrients, and washing out metabolites of anaerobiosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


