Fig. 19.1
Jamieson surgical classification of chronic thromboembolic disease (Reprinted from [36] with permission from Elsevier)
The surgical technique of sequential PTE with circulatory arrest was developed and standardized at the University of California San Diego (UCSD) and requires the use of cardiopulmonary bypass or extracorporeal circulation under conditions of deep hypothermia at 20 °C with briefs periods of circulatory arrest (limited to 20 min on each side). The procedure can be performed safely in patients with previous interventions including patients who have undergone previous coronary artery bypass grafting with patent bypass grafts [34].
The most relevant perioperative complication associated with PTE is pulmonary edema in the early postoperative period. Edema occurs in up to 20 % of patients who undergo PTE, particularly in patients presenting with high postoperative cardiac output. Careful fluid administration and restriction, reduction of cardiac output, and diuretics are important components of the perioperative management in patients with edema after PTE. Pulmonary hemorrhage is seen in up to 3 % of patients and is frequently associated with technical complications resulting in disruption of the vasculature during the endarterectomy, especially in cases with high PVR. Patients with pulmonary hemorrhage may require the use of postoperative ECMO support to allow stabilization. The most feared complication is the absence of surgically removable disease (Type IV); this may be associated with unchanged PVR and postoperative progression of RV dysfunction and is associated with poor early outcome [46].
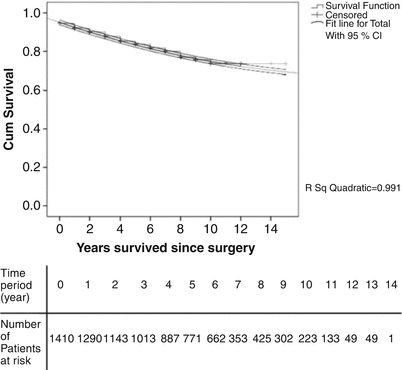
PTE is a curative procedure with excellent long-term outcomes. A systematic review demonstrated 5-year survival ranging from 74 to 89 % [47]. A recent analysis of the UCSD experience in more than 1400 patients followed longitudinally found that PTE was associated with a 10-year survival close to 70 % [33] (Fig. 19.2). Seventy-five percent of the patients who underwent PTE presented with NYHA functional class III–IV heart failure; almost 90 % of these patients were in functional class I–II at follow-up after intervention [35]. Despite these clinical and survival improvements, a subset of patients (10–30 %) will present with residual PH. Recently, a phase 3 study investigated the efficacy and side-effect profile of riociguat (a soluble guanylate cyclase stimulator) in patients with CTEPH who were ineligible for surgery or who had persistent or recurrent PH after PTE [48]. The study demonstrated hemodynamic and functional (6MWT) improvement, stimulating the use of medical management in patients with recurrent PH after PTE and patients ineligible for surgery.
Atrial Septostomy
The creation of an atrial septal defect without a thoracotomy using catheter and balloon-based techniques for patients with complete transposition of the great arteries was first introduced by Rashkind and Miller in 1966 and was then modified using blade septostomy by Park and colleagues in 1978 [49–51]. The first application of an atrial septostomy (AS) in patient with PAH was described by Rich and Lam in 1983 in a 22-year old woman with severe PH and RHF who was unresponsive to medical treatment with vasodilators. An AS was created percutaneously to offload her failing heart. The procedure was successful, but the patient died the following day due pulmonary edema and refractory hypoxemia [52]. Subsequently, the technique underwent several refinements. Although initial attempts were unsuccessful, they were followed by series demonstrating the efficacy of the technique with clinical and hemodynamic improvement (Fig. 19.3) [53, 54].
Fig. 19.3
Balloon dilators used for atrial septostomy
AS is based on the concept that survival of patients with PAH is closely correlated with the functional status of the RV, and that RV failure and syncope are associated with poor short-term outcomes [12, 55–57]. This rationale for AS is supported by the observation that patients with PAH with a patent foramen ovale (PFO) have better survival as compared with those who lack a PFO [58]. It has also been established that patients with Eisenmenger syndrome (a condition in which a congenital shunt and elevated pulmonary pressures result in deoxygenated blood in the left ventricle) have improved survival as compared with patients with IPAH, with a 3-year survival of 77 % vs. 35 % due to better cardiac output and lower RA pressures, despite higher pulmonary artery pressures [59]. Experimental work by Austen and colleagues in a canine model of PH showed that the creation of an AS decompressed the RV and increased the cardiac output on exertion [60].
The deterioration of the patient’s clinical condition and ultimately mortality caused by PAH are associated with the progressive dilation and dysfunction of the RV and the coexistent decrease of systemic output. The main predictors for decreased survival in PH are high RA pressure, high pulmonary artery pressure, and a low cardiac index [12, 56]. Creation of an AS allows a right-to-left arterial shunt, increasing the systemic cardiac output and oxygen transport to the periphery, despite a decrease in SaO2 (systemic arterial oxygen saturation), while allowing simultaneous decompression of the RV and improvement of RV function.
AS was initially performed using a blade balloon (BB) technique [53, 54]. Subsequently, the graded balloon dilatation (GBD) technique was popularized. The GBD technique provides more control over the size of the septostomy, with similar clinical and hemodynamic results as BB septostomy and a possible reduction of periprocedural risks [57, 61–63]. However, higher rate of spontaneous closure has been reported with the GBD technique [57]. Other techniques of creating a right-to-left shunt have been described that are based on the creation of a shunt between the descending aorta and the left pulmonary artery. These include the surgical creation of a Pott’s shunt (an anastomosis between the descending aorta and the left pulmonary artery), percutaneous creation of an aortopulmonary shunt, and percutaneous placement of a shunt following dilatation of a small probe-patent ductus arteriosus [64]. None of these techniques have been widely accepted. Most recently, mechanical cardiopulmonary support, including ECMO and Novalung, has been used in association with AS in decompensated patients with PH as bridge to lung transplantation, though the rapid and complete hemodynamic and respiratory support provided by ECMO and Novalung is decreasing the use of AS in patients with PH as a population [65].
The indications for AS in patients with PH lack strong evidence and are based primarily on limited single-institution series [61–63, 66]. The benefits and risks of AS were reported in an analysis of 64 cases published in the literature [67]. The main indication for AS is persistent RHF or recurrent syncope despite maximal medical therapy. Due to high periprocedural mortality in high-risk patients, AS should be avoided in patients in extremis. More recently AS has been used in patients with PH as a bridge to lung transplantation [63, 68]. AS has also been associated with the use of VV ECMO as a bridge to lung transplantation to avoid the thromboembolic and vascular complications present with VA ECMO [69, 70]. Occasionally, AS can be used as palliation, if lung transplantation is not an option.
Prior to the AS procedure, the candidate should have an adequate oxygen saturation (saturation of peripheral oxygen (SpO2) > 90 %) on room air or relatively low supplemental oxygen) to avoid profound desaturation once the right-to-left shunt is created. A hematocrit >35 % should be present to assure adequate oxygen-carrying capacity. Left ventricular (LV) function should be sufficient to tolerate the sudden in increase in blood return and volume overload [56, 57]. A left ventricular ejection fraction (LVEF) of >45 %, defined by echocardiogram, is preferable, and preconditioning patients with severe LV or RV dysfunction using inotropes and diuretics is recommended. Although severe RV failure remains a contraindication to AS, improving RV filling pressure using inotropes may allow the procedure [57]. Furthermore, the size of the septostomy should be carefully tailored. An AS that is too large can cause excessive shunting of blood away from pulmonary circulation, inadequate oxygenation, and excessive systemic desaturation. The excessive blood flow to the LV can precipitate LV failure and pulmonary edema further exacerbating desaturation and hypoxemia [57]. AS should be avoided in patients with left ventricular end diastolic pressure (LVEDP) >12 mmHg [52, 71]. Precautions for undertaking AS are outlined in Table 19.1.
Table 19.1
Recommendations for minimizing procedure-related mortality of atrial septostomy
Criteria | Recommendation |
|---|---|
Location | Only perform in a center experienced in pulmonary hypertension |
Actions to take before the procedure | Optimize cardiac function with adequate right heart filling pressure and additional inotropic support if needed Optimize oxygen delivery to maintain adequate hemoglobin (>15) with transfusion of packed red blood cells or darbepoetin alfa |
Actions to take during the procedure | Monitor variables (LAP, SaO2%, and mRAP) Tailor the defect to <10 % decrease in O2 saturation |
Actions to take after the procedure | Optimize oxygen delivery to maintain adequate hemoglobin (>15 g/dL) with transfusion of packed red blood cells or darbepoetin alfa |
Contraindications to atrial septostomy | Avoid atrial septostomy if the following contraindications are present: |
Severe right ventricular failure on cardiorespiratory support | |
mRAP >20 mmHg | |
PVRI >55 U/m2 | |
Resting O2 saturation <90 % on room air | |
LVEDP >18 mmHg |
The technique utilized to perform the AS (BB or GBD) will depend on the surgeon’s experience. Both techniques follow the same catheterization protocols using the left femoral artery and vein, followed by simultaneous right- and left-heart catheterization. The BB technique utilizes the Park blade. The GBD technique initially punctures the interatrial septum with a Brockenbrough needle followed with a stepwise, transseptal balloon dilation, beginning at 4 mm using the Inoue dilator followed by successive balloon dilation to 8, 12, and 16 mm. The final size of the AS is determined based on any of occurrence of (a) an increase of LVEDP to 18 mmHg, (b) reduction of SaO2 to ~80 %, (c) 10 % change from baseline or (d) diameter of 16 mm. This criteria is used to decrease the risk of excessive LV overload and refractory hypoxemia, which was identified as the most important cause of mortality in many studies [52, 67, 71]. Intraprocedural and postprocedural echocardiograms are useful to assess changes in the LV and RV and to determine final size of the AS, typically about 80 % of the diameter of the largest balloon utilized [52, 67, 71].
Limited single-institution series have provided insight into the immediate-term and long-term effects of AS in patients with PH. Sandoval and colleagues described the creation of AS in 15 patients with severe PPH with mean PA pressure of 66 ± 13 mmHg. They defined the basic requirements of SaO2 > 80 %, hematocrit >35 %, and reasonable RV function before undergoing AS. AS was created percutaneously and dilated progressively to 8–16 mm, keeping SaO2 > 75 % and LVEDP <18 mmHg. Postprocedure, there was an immediate and significant decrease in RV end-diastolic pressure and SaO2 and an increase in cardiac index. One patient died, and 14 survived the procedure and significantly improved their mean NYHA functional class and 6MWT [57]. Similar improvements in cardiac index and mid-term survival were observed by others [56]. In the first overall analysis of the worldwide experience with AS, the early postprocedural mortality after AS in patients with PH was 16 %, but implementation of the guidelines to minimize the procedure-related mortality of AS (Table 19.1) decreased this rate to 5.4 % [46, 67]. The immediate effects of the AS on resting pulmonary hemodynamics were variable. The mean RA pressure declined by 3 mmHg; the cardiac index increased by 0.7 l/min/m2. Baseline mean RA pressure is the best indicator of hemodynamic response, and the best hemodynamic and clinical responses are obtained when mean RA pressure is between 10 and 20 mmHg. Patients with mean RA pressure > 20 mmHg may also present adequate hemodynamic response but with a higher risk of mortality [57, 67].
The long-term results of the AS have not been well documented. Kerstein and colleagues noticed an improvement in the RV function with improvement in diastolic PAP and cardiac index in patients studied 7–27 months after AS [53]. Long-term clinical response seems to have a close correlation with the immediate response including cardiac index and oxygen transport [63]. In this study, patients with greater increases in cardiac index (52 % vs 15 %) and systemic oxygen transport (37 % vs 4 %) shortly after the AS procedure had an improved clinical response. The median long-term survival of 54 patients surviving AS was 19.5 months. Progression of the underlying pulmonary vascular disease was the main factor associated with late mortality.
Transplantation
Lung transplantation is an established therapeutic option for patients with advanced respiratory failure caused by many etiologies including restrictive and obstructive diseases, infectious complications, and pulmonary vascular complications as in the case of PAH. The first lung transplantation in a human was performed by Hardy in 1963 in a patient with advanced lung cancer who died soon after the intervention due to renal failure [73]. The first heart-lung transplant was successfully performed at Stanford University by Norman Shumway, John Wallwork, and Bruce Reitz in 1981 for PAH [74]. Joel Cooper reported the first successful single lung transplant for pulmonary fibrosis in 1983, followed by the first series of bilateral lung transplants in 1986 [75–77]. Since 1995, more than 47,000 lung transplants and 3700 heart-lung transplants have been performed [78, 79].
Although PAH was recognized as a clinical entity in the early 1950s, the medical community lacked effective treatments for decades, with a median survival of only 2.8 years [12, 80]. Thirty years later, the treatment and prognosis of patients with PAH was notably improved with the introduction of heart-lung transplantation and subsequently lung transplantation as a valid treatment alternative. Due to technical considerations and concerns regarding postoperative RV performance, heart-lung transplantation was considered the procedure of choice in patients with PAH, whereas lung transplantation was used mostly for other forms of end-stage lung disease [81]. After the efficiency of prostacyclin analogs, endothelin receptor antagonists, and PDE5 inhibitors became evident, transplantation remained a second-line treatment for years. As medical therapy improved, we observed an important decrease in pulmonary vascular disease as indication for transplantation—representing 10–15 % of patient undergoing transplantation initially but only 4 % in more contemporary reports [78, 79]. Most recently, with improvements in surgical techniques and outcomes in lung transplantation and the understanding that despite optimal medical therapy up to 30 % of patients will progress rapidly or respond poorly to medical treatment, transplantation has regained attention in the management of PAH [81, 82].
Indication and Timing for Transplantation
The indications and patient selection for lung or heart-lung transplantation have been previously stated by International Society for Heart and Lung Transplantation (ISHLT) consensus [83]. In general, patients are considered for transplant if they have advanced end-stage lung disease, NYHA functional class III–IV heart failure, have a life expectancy of < 2 years, and have no contraindications [83]. The indications for lung transplantation referral in patients with PH are summarized in Table 19.2.
Table 19.2
ISHLT guidelines for transplantation referral
1. NYHA functional class III or IV, on maximal medical therapy |
2. Rapidly progressing disease with low (<350 m) or declining 6 min walk distance |
3. Syncope |
4. Additional risk factors for poor outcome in PAH |
(a) Hyponatremia |
(b) Hyperbilirubinemia |
5. Right ventricular dysfunction: |
(a) Cardiac index < 2 l/min/m2 |
(b) Right atrial pressure > 15 mmHg |
(c) Tricuspid annular plane systolic excursion <1.8 cm |
6. Underlying connective tissue disease (e.g. scleroderma) |
7. Chronic obstructive pulmonary disease |
8. Sarcoidosis |
9. Pulmonary fibrosis |
10. Combined emphysema, pulmonary fibrosis, and PAH |
In patients with PAH, an adequate response to medical treatment occurs in the majority of patients and may be responsible for a delay in referral for lung transplantation. However, there are other factors that need to be considered at the time of referral. The development of the lung allocation scoring (LAS) system, implemented in 2005 with the intention to increase access to lung transplantation in higher risk patients, may not accurately reflect risk in patients with IPAH [11, 84]. The LAS is a scoring system, developed from demographic and clinical characteristics, that takes into account waiting list urgency and probability of posttransplant survival [84]. After implementation of the LAS system, deaths while on the waiting list for a lung transplant decreased for all diagnosis groups except for patients with IPAH, which remained unchanged. Patients with IPAH were less likely receive a transplant than patients with idiopathic pulmonary fibrosis (IPF) or cystic fibrosis (CF) and more likely to die on the waiting list than patients with chronic obstructive pulmonary disease (COPD) or CF [83, 85, 86].
Some variables associated with worse prognosis in patients with PAH, as studied in the REVEAL cohort, are not represented in the LAS. These variables include right atrial pressure (RAP) >14 mmHg and 6MWT < 300 m [11]. Recent analyses have suggested that current prediction models that utilize the LAS may overestimate the survival of patients listed with IPAH. New statistical models that consider other variables, including 6MWT, resting O2, invasive cardiac output testing, and NYHA functional class, have been suggested to help guide clinical decision-making [11, 87]. For this reason, the United Network for Organ Sharing (UNOS) now considers LAS appeal in lung transplant candidates with PAH [82]. If certain criteria related to declining right ventricular function are met (including deterioration in clinical condition on optimal medical therapy, right atrial pressure >15 mmHg, and cardiac index <1.8 L/min/m2), an LAS is assigned such that the patient is placed in the 90th percentile on the wait list.
Debate still exists regarding outcomes on the wait list and posttransplantation survival after implementation of the LAS score. Schaffer and colleagues in a recent review of the UNOS registry found that after LAS implementation, the incidence of transplantation increased and wait-list mortality decreased in patients with IPAH, and that both heart-lung transplantation and lung transplantation are predictive of survival, as is being listed at a medium- to high-volume center [88]. This report provided a positive perspective of the problem. Unfortunately, we continue to see patients coming late for referral for transplantation, and in some circumstances, they are not able to be considered for a transplant due to their advanced disease and multi-organ involvement. Patients with PAH who are listed continue to have a high mortality while on the wait list—as high as 34 % in recent analyses by UNOS and others [89]. This has forced experienced transplant centers to suggest early referral of patients with PH for transplant evaluation. Despite the availability of advanced circulatory support systems, including ECMO and Novalung, to bridge patients to transplantation, advanced circulatory support is only possible in selected and experienced centers [90–96]. It does not seem appropriate to wait until advanced right-sided heart failure, liver failure, frequent arrhythmias, and syncope are present to consider referral.
Lung transplantation generally is offered to patients with PH who do not respond to optimal vasodilator therapy [82]. Updated treatment algorithms have been proposed in patients with PAH in NYHA class IV [97]. In an attempt to prevent late referral for lung transplantation, some experienced centers consider initiation of treatment with intravenous epoprostenol to be a better time to perform an initial lung transplantation evaluation [89]. Due to the difficulty in predicting the clinical course of patients with PAH and associated PAH (APAH; PAH resulting from another disease), early referral is recommended even for a preliminary evaluation. The inadequate response of a patient with PAH to monotherapy should prompt physicians to consider a lung transplantation evaluation in parallel with initiation of sequential combination therapy. Failure of the combination therapy should then trigger the activation of the patient on the transplant wait list [97].
Sitbon and colleagues identified a high-risk subset of patients with PH who are likely to have a poor outcome without transplantation. These include patients with a history of right-sided heart failure, who remained in NYHA functional class III or IV after 3 months on epoprostenol, who showed a 6MWT <250 m, and who had high RAP (>12 mmHg) along with the absence of a decrease in total pulmonary resistance of >30 %, relative to baseline. These patients should be prioritized for lung transplantation [8].
It is worth mentioning that patients with PH associated with underlying lung disease, such as COPD, pulmonary fibrosis, or sarcoidosis, are also at increased risk of mortality, and patients with combined emphysema, interstitial lung disease and PH have a particularly high mortality risk with 1-year survival in only 60 % [98–101]. In these patients with lung disease and PH, the variable effects of oral and inhaled vasodilators with the potential to worsen symptoms frequently indicate the need of an expedited lung transplantation evaluation [82].
Considering the implications of severe PAH in the management and prognosis of patients being considered for transplantation, it is advisable that these patients are cared for during the mid-late phase of the clinical course of their disease by an experienced and multidisciplinary group with the ability to provide lung and heart-lung transplantation, mechanical respiratory and circulatory support, and other temporizing strategies.
Type of Transplantation
With the utilization of cyclosporine and better immunosuppressive regimens beginning in the early 1980s, the survival of patients after heart-lung transplantation improved markedly [102, 103], and heart-lung transplantation became the accepted treatment for patients with advanced PAH and other advanced cardiopulmonary diseases. The acceptable outcomes of various single-center experiences were published [81, 104, 105]. PAH remains an important indication for heart-lung transplantation with 27.6 % of heart-lung transplants being performed for PAH, second in frequency only to heart-lung transplantation performed for cyanotic congenital heart disease [106]. As the overall number of patients listed for transplantation increased and the organ scarcity became evident, single-lung transplantation and double-lung transplantation were considered as alternatives to heart-lung transplantation in patients with PH. More recently, double-lung transplantation has been favored over single-lung transplantation [81, 107–109].
In the 1990s, our group at the University of Pittsburgh demonstrated that single-lung transplantation could be done with survival comparable with double-lung transplantation [107, 108], suggesting that single-lung transplantation could be considered the procedure of choice in patients with PAH to increase organ utilization. Gammie and co-workers reviewed the results of 58 lung transplants performed for PPH or secondary PH; 37 double-lung transplants and 21 single-lung transplants were performed. As expected, cardiopulmonary bypass times were longer in patients who underwent double-lung transplantation as compared with those who underwent single-lung transplantation. Recipients with PH had similar functional status and similar early- and mid-term survival after single-lung transplantation or double-lung transplantation [109]. We have recently observed patients surviving more than 20 years after single-lung transplant who occasionally require redo transplantation.
A recent report from the Cleveland Clinic concluded that the use of single-lung transplantation in older patients (>65 years) with secondary PH was associated with similar outcomes as double-lung transplantation. Murthy and colleagues presented a series of single-lung transplant recipients that compared 36 patients with PH (mean PAP 30.4 mmHg) and 48 patients with no PH. Survival at 3 years was similar, 70 % and 72 % respectively, with patients with PH having only a longer hospital stay (24 days vs 16 days; p = 0.015). This study suggested that single-lung transplantation could be an acceptable solution to minimize the surgical intervention and increase organ utilization [110].
Other series have associated double-lung transplantation with better outcomes than single-lung transplantation, however, and perioperative management of patients after double-lung transplant may be more straightforward. In a series of patients with PH, including 27 patients with PAH and 30 patients with Eisenmenger syndrome, Bando and co-workers from our institution found that double-lung transplantation and heart-lung transplantation provided a better option than single-lung transplantation in reducing pulmonary arterial pressure, improving cardiac index, and providing better functional outcomes and better early and mid-term survival [107, 108]. Subsequently, Conte and colleagues, in a study that included patients with PPH (n = 15) and secondary PH (n = 40), observed a significant improvement in survival in patients with PPH who underwent double-lung transplantation as compared with single-lung transplantation in patients with PPH as the indication for transplantation [111].
The combined clinical experience of many groups has demonstrated the complexity of the perioperative management needed for single-lung transplantation because the majority of the cardiac output (80–90 %) perfuses the newly implanted lung. There is an increased likelihood of needing ECMO support due to the increased risk of severe primary graft dysfunction (PGD) in patients with PH [106, 112]. Using double-lung transplantation may also eliminate the risk of recurrent PH seen occasionally after single-lung transplantation [81]. Thus, double-lung transplantation may be a safest alternative. Since this realization, the use of single-lung transplantation has decreased rapidly with fewer than 5 % of patients with PAH undergoing single-lung transplantation, as per ISHLT transplantation registry reports [106, 112].
Double-lung transplantation is currently the preferred choice worldwide over heart-lung transplantation for patients with PH, especially considering the importance of organ scarcity. Double-lung transplantation offers comparable outcomes as heart-lung transplantation in patients with PAH with the advantage of better organ sharing [113]. A recent meta-analysis used nine selected studies to answer the question, “Should we perform double-lung transplantation or heart-lung transplantation in patients with PH?” Reviewing 1189 lung transplants (67 %) and 578 heart-lung transplants, the authors concluded that neither procedure had an overall survival benefit over the other [113]. Interestingly, the same study corroborated that heart-lung transplantation is associated with excellent functional and survival outcomes in patients with congenital heart disease and Eisenmenger syndrome, severe right ventricular dysfunction (EF < 10–25 %) or left ventricular dysfunction (EF < 35–55 %), and those who are chronically inotrope-dependent. In all other patients with PH, double-lung transplantation offered similar outcomes with better organ utilization [113]. Supporting the use of heart-lung transplantation in the situations mentioned above, some authors have reported a lower PGD rate and better survival without bronchiolitis obliterans syndrome (BOS) when heart-lung transplantation was performed [81]. Dartevelle and colleagues reviewed their experience with lung transplantation and heart-lung transplantation in patients with severe PH with an extensive analysis of 219 patients: 152 who underwent heart-lung transplantation and 67 who underwent lung transplantation for end-stage PH (including 147 patients with PAH) over a 22-year period (1986–2008). They concluded that in patients with end-stage PH, good long-term outcomes were obtained with both protocols as indicated by a 43 % survival after double-lung transplantation and 39 % survival after heart-lung transplantation at 10 years posttransplant (p = 0.9) (Fig. 19.4). These similar survival outcomes were obtained despite the fact that the heart-lung transplantation group was composed of sicker patients with worse NYHA functional class heart failure, worse kidney and liver function, and a higher need for preoperative inotropes as compared with the double-lung transplantation group [81]. Heart-lung transplantation was associated with lower postoperative morbidity, shorter MV time, lower rates of bronchial complications and postoperative PGD, and greater freedom from BOS-related death at follow-up. Heart-lung transplantation has been associated with a certain degree of immunologic protection to the lung, which the authors put forth to explain the lower rate of BOS after heart-lung transplantation as compared with double-lung transplantation in this study [81]. This theory has not been supported by other studies [114–116].
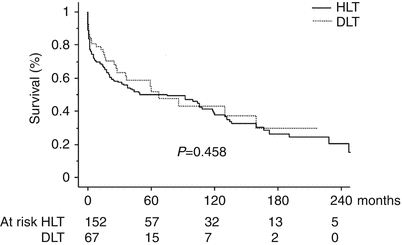
Fig. 19.4
Outcomes of double-lung transplantation vs heart-lung transplantation. DLT double-lung transplant, HLT heart-lung transplant (Reprinted from [81] with permission by Oxford University Press)
Heart-lung transplantation continues to be an excellent alternative in patients with a complex congenital systemic-to-pulmonary shunt (e.g. Eisenmenger syndrome), with intra-cardiac lesions that require complex repair (e.g. a ventricular septal defect), or with profound RV dysfunction and dilation. In an analysis of transplantation for Eisenmenger syndrome, survival was significantly better after heart-lung transplantation than after lung transplantation, a difference that was not apparent for recipients with IPAH [117]. This benefit was more pronounced when the congenital defect was a ventricular septal defect (VSD).
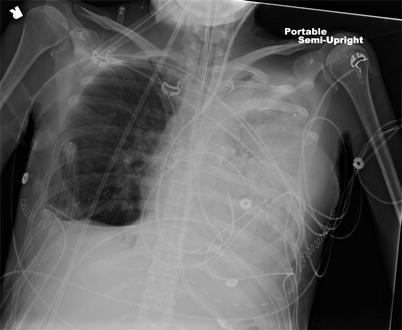
Although double-lung transplantation may be doable in the majority of patients, even those with severe RV dysfunction, a careful consideration should be made in patients with extreme cardiac dilation, as severe compression of the vascular structures on the left-side of the chest may occur as a consequence of cardiac-left chest cavity mismatch or cardiac herniation, leading occasionally to a loss of the allograft requiring redo transplantation [118]. We have experienced a similar situation following double-lung transplantation (Fig. 19.5).