Danon disease is an X-linked systemic disorder characterized by left ventricular hypertrophy, mental retardation, and skeletal myopathy affecting young men. Electrocardiogram usually displays a Wolff–Parkinson–White preexcitation pattern. Less has been reported about the phenotype in women, although later-onset cardiac symptoms have been described. The aim of this study was to expand the knowledge of the phenotype of Danon disease in women. We clinically followed and evaluated with echocardiography, cardiac magnetic resonance imaging (cMRI), and genetic testing a family affected by Danon disease in which 2 men and 6 women showed a severe arrhythmogenic phenotype. Affected family members carried a nucleotide substitution at position 294 in exon 3 (c.294 G → A) that changed a tryptophan residue to a stop codon at position W98X in the lysosome-associated membrane protein 2 ( LAMP2 ) gene. Four women died suddenly (1 aborted) at 37 to 54 years of age. Wolff–Parkinson–White pattern with atrioventricular block was detected in 2 of 6 women. Four had successful pregnancies without symptoms of heart failure. cMRI showed late gadolinium enhancement areas in a clinically healthy woman who was a mutation carrier. Two patients underwent heart transplantation; histology of explanted hearts demonstrated severe interstitial fibrosis, hypertrophic cardiomyocytes with cytoplasmic vacuoles, and myofibrillar disarray. In conclusion, LAMP2 mutation can cause a severe arrhythmogenic phenotype in women that includes a high risk of sudden death. cMRI may be useful in women harboring LAMP2 mutations to permit early detection of cardiac involvement and guide timely considerations of implantable cardioverter–defibrillator therapy. Heart transplantation should be considered at onset of heart failure symptoms owing to rapid progression of the disease.
Danon disease is a rare X-linked multisystemic disorder that has been most thoroughly described in boys. In the original report the disease was clinically characterized by hypertrophic cardiomyopathy, skeletal myopathy, and mental retardation in young men and was described as a lysosomal glycogen storage disease with normal acid maltase. Subsequently, it was determined that a deficiency of the lysosome-associated membrane protein 2 (LAMP2) caused the disease. Currently few data are available in women with Danon disease who are generally described as being at risk for cardiac involvement with milder and later onset of cardiac symptoms compared to men. Cardiac disease in women can present with a broad spectrum of clinical features that vary from hypertrophic to dilated cardiomyopathy. Preexcitation patterns on electrocardiogram occasionally can be the sole expression and marker of the disease in women. Recently, Toib et al described earlier cardiac involvement in women including dilated, hypertrophic, and peripartum cardiomyopathy. Here we report the unique phenotype of a large family with Danon disease in which women were characterized by an unusually severe arrhythmogenic trait and sudden cardiac death.
Methods
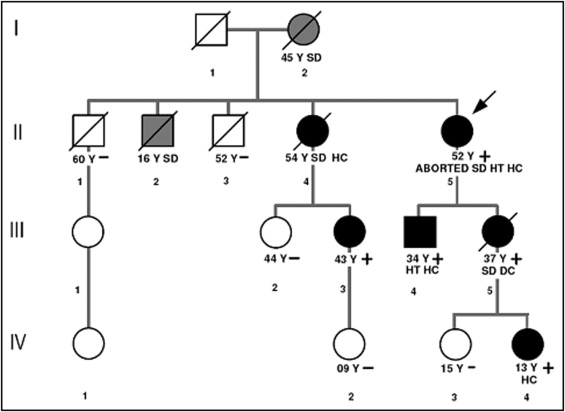
Five of 8 affected family members of a large kindred with Danon disease (4 women and 1 man) have been followed by the transplantation and heart failure outpatient clinic of the University Hospital S. Maria della Misericordia, Udine, Italy ( Figure 1 , Table 1 ). Of the remaining affected relatives, 1 (II-4) was followed by another center and died before coming to our observation; her medical records were reviewed. Two other members of this family (I-2 and II-2) died suddenly before examination.
| Case | Age at Onset (years)/Sex | Clinical Presentation | Medical Treatment | ECG | LVEF | Phenotype | ICD/PM | Outcome | Age (years) |
|---|---|---|---|---|---|---|---|---|---|
| II-5 | 25/F | Palpitations | Metoprolol, amiodarone, furosemide propafenone | WPW LVH | 56% | HC | ICD | Aborted SD/transplantation | 52 |
| II-4 | 24/F | Palpitations | Amiodarone | WPW LVH | Normal | HC | PM | SD | 54 |
| III-4 | 19/M | Palpitations, exertional dyspnea, heart failure | Nebivolol, captopril, furosemide | WPW LVH | 60% | HC | ICD | Transplantation | 34 |
| III-3 | 36/F | Palpitations | Bisoprolol | Normal | 61% | ICD | ICD | 43 | |
| III-5 | 36/F | Dyspnea, fatigue | Bisoprolol, enalapril, furosemide | Incomplete LBBB | 32% | DC | — | SD | 37 |
| IV-4 | 13/F | Palpitations | None | LVH | 79% | HC | ICD | ICD | 15 |
All living patients were evaluated by clinical history, physical examination, electrocardiography, echocardiography (M-mode, 2- and 3-dimensional, and Doppler), and Holter monitoring; cardiac magnetic resonance imaging (cMRI) with late gadolinium enhancement was performed in 2 patients. Dilated cardiomyopathy was defined as left ventricular ejection fraction (LVEF) <45% and LV hypertrophy was defined as LV septum or posterior wall >2 SDs above the normal limit for patients ≤18 years of age or 15 mm for patients >18 years of age. Creatine kinase levels were obtained in all living patients. Diagnosis of Danon disease was based on a combination of DNA analysis, immunohistochemistry, and family history in each case. Mean follow-up was 55 months (range 11 to 114). All living patients provided informed consent for the study according to the institutional review committee.
LAMP2 genetic analysis was performed in 8 symptomatic or at-risk family members ( Figure 1 ) by extracting genomic DNA from blood leukocytes or muscle biopsy using the GenElute Mammalian Genomic DNA kit (Sigma, St. Louis, Missouri). The entire coding sequence of the LAMP2 gene was amplified in 10 amplicons using primer sequences (available on request) designed using the human LAMP2 sequence as reference ( NM_002294.2 ). Polymerase chain reactions were performed under standard conditions. Polymerase chain reaction products were purified by enzyme reaction (ExoSAP-I, Amersham, Buckinghamshire, United Kingdom), quantified on agarose gel, and directly sequenced using the Big Dye di-deoxy-terminator cycle sequencing kit and the 377 ABI-PRISM automated sequencer (Applied Biosystems, Foster City, California) at the Innovative Biotechnology Center, University of Padua (Padua, Italy).
Results
The proband (II-5), a woman, developed symptoms of palpitations at 25 years of age. Her electrocardiogram showed a Wolff–Parkinson–White (WPW) pattern ( Figure 2 ), LV hypertrophy, and paroxysmal atrial flutter and fibrillation. Subsequently an echocardiogram showed LV hypertrophy with EF 56%. The patient had hearing loss, myopia, and peripheral myopathy with normal creatine kinase levels. By 50 years of age she was symptomatic for advanced heart failure and developed several syncopal episodes. Echocardiogram showed LVEF 33%. During hospitalization for cholecystectomy, she had ventricular fibrillation and was successfully resuscitated and treated with an implantable cardioverter–defibrillator (ICD). She underwent urgent heart transplantation at 52 years of age. Genetic analysis demonstrated a LAMP2 mutation (c.294 G → A) predicting a typical Danon disease nonsense mutation (W98X).
The proband’s mother (I-2) was never formally diagnosed with heart failure or arrhythmia, although she is suspected to have carried the mutation because ≥2 of her children inherited the mutation and she died suddenly and unexpectedly at home at 45 years of age; no autopsy examination was performed.
Patient II-4 developed heart palpitations at 24 years of age, and electrocardiogram obtained at the same time showed severe hypertrophy and WPW pattern with atrial flutter. Later she developed advanced atrioventricular block and a pacemaker was implanted. Echocardiogram showed LV hypertrophy and normal LVEF. She died suddenly at 54 years of age.
Patient III-3 was followed clinically since 36 years of age after testing positive for the LAMP2 mutation. She complained of palpitations and cMRI showed clear late gadolinium enhancement in the inferior wall with a central myocardial pattern, although repeated echocardiograms were normal. Holter monitoring showed frequent complex ventricular arrhythmias (>30/hour) and nonsustained ventricular tachycardia. Ophthalmologic evaluation found a small area of retinal epithelium dystrophy. She received an ICD as primary prevention because of a family history of sudden death and had successful discharge of her ICD to interrupt a sustained fast ventricular tachycardia.
The proband’s daughter (III-5) underwent genetic testing at 33 years of age and showed the LAMP2 mutation when she had no cardiac symptoms. She developed dyspnea at 36 years and was noted to have a mildly dilated cardiomyopathy with LVEF 32% ( Figure 3 ). After optimal medical therapy for heart failure, she recovered and echocardiographic EF was 50% after 4 months. She refused an ICD and died suddenly at 37 years of age.



