Fig. 27.1
Onset of polymorphic VT symptomatic for syncope, converted to sinus rhythm after cardiopulmonary resuscitation
Medical History and Cardiovascular Risk Factors
Ebstein anomaly and severe pulmonary stenosis; her family history of structural heart disease, syncope, or sudden cardiac death was unremarkable.
2014: She had undergone surgical tricuspid valve replacement with a biological prosthetic valve and cavopulmonary anastomosis.
Allergies
None
Medications
None
Vital Signs
Temperature: 36.8 °C
Heart rate: 80 bpm
Arterial blood pressure: 110/70 mmHg
Respiratory rate: 18 breaths/min
Oxygen saturation: 92 %
Physical Examination
General appearance: Well developed, well nourished, alert, and cooperative
Lungs: Clear to auscultation and percussion without rales, rhonchi, wheezing, or diminished breath sounds
Cardiovascular: Normal S1 and S2. No S3 and S4. Systolic murmur 2/6 at Erb’s point. Regular rhythm. No peripheral edema, cyanosis, or pallor. Warm and well-perfused extremities
Abdomen: Positive bowel sounds. Soft, nondistended. No guarding or rebound. No masses
Routine Laboratory Test
Tests were normal (hemoglobin 12.8 g/dl; white blood cells 9680/mmc; creatinine 0.82 mg/dl; potassium 3.7 mEq/l; sodium 138 mEq/l; magnesium 1.7 mg/dl).
Instrumental Examination
Basal ECG after DC shock was normal with incomplete right bundle branch block and specific abnormalities of repolarization.
The loss of consciousness was caused by a monomorphic pulseless VT (Fig. 27.1), converted to sinus rhythm by external DC shock after a brief period of cardiopulmonary resuscitation.
Figure 27.2 shows the magnetic resonance of the patient. The right ventricle was severely enlarged, with a papyraceous right ventricular wall.
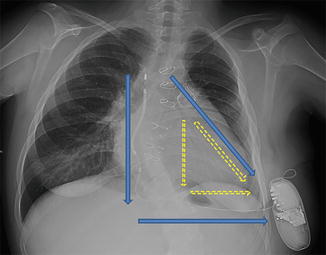
Fig. 27.2
Cardiac magnetic resonance
Clinical Course and Therapeutic Management
During hospital stay, she was treated with a beta-blocker (propranolol 3 mg/kg/day) with no further episodes. Genetical analysis and exercise test ruled out polymorphic ventricular tachycardia as the etiology of the VT.
The VT episode was treated with an external direct current cardioversion, and the patient was then referred to our center to attempt a VT ablation. Unfortunately, the fact that the right ventricle (RV) was papyraceous and the free ventricular wall was thin would have exposed the patient to a high risk of catheter perforation.
Because transvenous implantable cardioverter defibrillator (ICD) implantation had the same risk of cardiac perforation and access to the right cardiac chambers had been made unfeasible by cavopulmonary anastomosis, subcutaneous (S)-ICD implantation was considered. In assessing the patient’s adequacy for S-ICD implantation, the morphology of the QRS was screened to avoid T-wave oversensing. Because of the bizarre morphology of the QRS attributable to severe dilatation of the RV, QRS screening was performed with the left arm electrode placed to the right side of the xiphoid process. Under local anesthesia and conscious sedation, the catheter was inserted subcutaneously from the pocket in the left midaxillary region to the right side of the xiphoid process. The tip was then advanced up to the manubriosternal junction, 1 cm to the right of the midsternal line (Fig. 27.3). At the end of the procedure, a defibrillation test was performed: Clinical VT was induced, correctly sensed, and treated after 12.5 s with a 65 J shock (conventional polarity configuration). At 2-month follow-up examination, the patient did not have any complications, and no significant events were recorded.
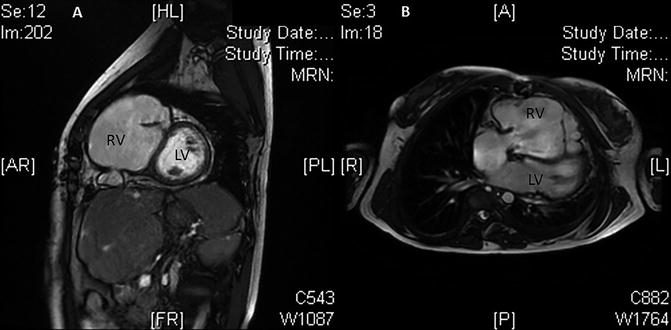
Fig. 27.3
Chest X-ray of S-ICD implant
27.3 Sudden Cardiac Death
Definitions
Various criteria have been used to define SCA and SCD in the medical literature [3]. It is difficult to find a specific definition for several main reasons: First of all, only two-thirds of events are witnessed, making the diagnosis difficult to establish; secondly, the cardiac rhythm at clinical presentation is unknown in many cases, so it is not possible to restrict the definition of SCA to documented cases of VF; finally, the suddenness of death is generally defined by the duration of symptoms prior to SCA, but in approximately one-third of cases, the duration of symptoms is not clearly defined.
For these reasons, the criteria proposed do not rely upon the cardiac rhythm at the time of the event. The criteria focus on the sudden pulseless event occurred out of hospital and on the absence of a noncardiac condition (e.g., pulmonary embolism, intracranial hemorrhage, central airway obstruction) as the cause of cardiac arrest.
The following definitions of SCA and SCD were presented by the 2006 American College of Cardiology/American Heart Association/Heart Rhythm Society (ACC/AHA/HRS):
Sudden cardiac arrest is the sudden cessation of cardiac activity so that the victim becomes unresponsive, with no normal breathing and no signs of circulation. If corrective measures are not taken rapidly, this condition progresses to sudden death. Cardiac arrest should be used to signify an event as described above, that is reversed, usually by CPR and/or defibrillation or cardioversion, or cardiac pacing. Sudden cardiac death should not be used to describe events that are not fatal [1].
Epidemiology
SCD accounts for approximately 15 % of the total mortality in the industrialized countries [4].
The incidence of SCD increases as a function of advancing age. The prognosis of patients experiencing SCA varies significantly according to the initial rhythm, although the outcome remains poor, despite advances in the treatment of heart disease.
The incidence of experiencing SCA is increased by age, sex, and underlying cardiac disease [5]. Indeed, men are two to three times more likely to experience SCA than women. In the Women’s Health Initiative, 161,808 postmenopausal women were followed for an average of 10.8 years, and the incidence rate of SCD was 2.4 per 10,000 women/year, and nearly one-half of women who experienced SCD did not have prior clinically recognized coronary heart disease [6].
The presence of clinically recognized heart disease and of coronary heart disease (CHD) risk factors, respectively, increases six- to tenfold and two- to fourfold the risk of SCA [7].
Etiology
SCA is more frequent in male with structural heart disease, especially CHD. The most frequent cause of SCA is CHD that amounts up to 70 % of SCAs. It is possible that SCA occur during an acute coronary syndrome (ACS) such as in patients with a chronic, stable CHD [8]. Usually in this setting, prior myocardial damage and scar act as a substrate for SCA. On the contrary, SCD occurs more commonly in the absence of an identifiable acute cardiac event.
Ten percent of cases of out-of-hospital SCA is represented by other forms of structural heart disease, both acquired and hereditary. Examples of such disorders include the following:
1.
SCD is responsible for approximately one-third of deaths in heart failure and cardiomyopathy.
2.
Left ventricular hypertrophy due to hypertension or other causes.
3.
Myocarditis.
4.
Hypertrophic cardiomyopathy.
5.
Arrhythmogenic right ventricular cardiomyopathy.
6.
Congenital coronary artery anomalies.
7.
Mitral valve prolapse.
Data from different reports documented approximately 10–12 % of cases of SCA among subjects without structural heart disease, under age 45, while a lower value of about 5 % has been described when older patients are included [9, 10].
Hereditary factors that contribute to coronary heart disease risk have been thought to operate nonspecifically for the SCD syndrome. However, several studies have identified mutations and relevant polymorphisms along the cascade from atherogenesis to plaque destabilization, thrombosis, and arrhythmogenesis, each of which is associated with a risk of a coronary event.
SCA in the absence of apparent structural heart disease can occur in different settings:
1.
Brugada syndrome
2.
Idiopathic VF
3.
Congenital or acquired long QT syndrome
4.
Arrhythmogenic right ventricular cardio-myopathy
5.
Catecholaminergic polymorphic VT
6.
Familial SCD of uncertain cause
7.
Wolff-Parkinson-White syndrome
A major role in the pathogenesis of SCA, in addition to the presence of the above underlying disorders, is played by acute superimposed triggers. These include ischemia, autonomic nervous system activation, psychosocial factors, electrolyte disturbances (particularly hypokalemia and hypomagnesemia), and the proarrhythmic effect of some antiarrhythmic drugs. The mortality is highest in the first month after acute myocardial infarction (MI) in patients with ejection fraction (EF) of less than 30 % [1].
In addition, direct trauma over precordium precipitated SCA that results from commotio cordis.
The cumulative risk of SCD has been estimated at 15–20 % of adults with aortic stenosis, with the risk being higher in symptomatic patients and equal to or less than 5 % in asymptomatic patients. Mitral valve prolapse is usually non-life-threatening, and its link with SCD has never been definitely demonstrated. Reported SCD rates in patients with Wolff-Parkinson-White (WPW) syndrome have been 0.15 %, due in most to the development of atrial fibrillation (AF) with a rapid ventricular response that degenerates to VF. Genetic influences modulate the risk of SCD in the setting of CHD. The Paris Prospective Study I, analyzing more than 7,000 men followed for an average of 23 years, found that a parental history of SCD increased the relative risk of SCD for offspring to 1.8, without elevating the risk for MI. When both parents had SCD, the relative risk for SCD in offspring was 9.4. Genetic influences may act through multiple mechanisms, such as modulation of the substrate, atherothrombosis, electrogenesis impulse propagation, neural control, and regulation [1].
Some individuals can have inherited abnormalities that are not manifest until triggered by an external event. For example, autonomic modulation associated with certain types of activity, as well as drugs that affect cardiac repolarization, can convert a subclinical genetic abnormality to SCD. Among the genetic factors, the most common are DNA variants called “polymorphisms” that may be present in a large proportion of the population and create susceptibility for SCD. Single nucleotide polymorphisms (SNPs) are DNA variants that can be associated with a functional consequence. For example, a polymorphism identified in the alpha 2b adrenergic receptor is associated with an increased risk of MI and SCD [1]. Nevertheless, to create a risk for SCD, a combination of polymorphisms in different genes may be required, interacting with a specific trigger or substrate, because millions of SNPs are present in the DNA of each individual [1].
Risk Factors
An increased risk of SCA is associated with a number of clinical characteristics and other factors among patients without prior clinically recognized heart disease [11, 12]. Coronary heart disease and SCA have a lot of risk factors in common. These include family history of myocardial infarction, hypertension, dyslipidemia, diabetes mellitus, obesity, cigarette smoking, and physical inactivity [13, 14].
The risk of SCA in patients with coronary heart disease is strongly related to current cigarette smoking and the number of cigarettes smoked per day. In the Nurses’ Health Study, 101,018 women were followed for 30 years, and current smokers had a significantly greater risk of SCD than women who had never smoked (adjusted hazard ratio 2.44, 95 % CI 1.80–3.31), and the risk was increased even among women who smoked 1 to 14 cigarettes per day (adjusted hazard ratio 1.84, 95 % CI 1.16–2.92) [15]. In this study, the risk of SCD declined over time in a linear fashion after quitting smoking, reaching the same risk of SCD as never smokers 20 years after quitting [15].
The risk of SCA is particularly high among current smokers and declines rapidly after stopping smoking, to reduce the risk of SCA and a multitude of other complications.
During and up to 30 min after strenuous exercise, the risk of SCA is transiently increased compared to other times [16]. In fact, the actual risk during any one episode of vigorous exercise is very low (1 per 1.51 million episodes of exercise) [17]. Also, the magnitude of the transient increase in risk during acute exercise is lower for people who perform regular exercise compared with people for whom exercise is unusual [16, 17].
Data from long-term exercise intervention trials among apparently healthy persons that focus upon major disease end points are absent. Anyway, regular exercise should be encouraged for the primary prevention of coronary heart disease and consequently SCA. Despite a small transient increase in risk during and shortly after strenuous exercise, rate of SCD is lower among exercisers compared with sedentary men [17]. A reduced risk of SCD is associated with a lower resting heart rate and increased heart rate variability, characteristics which developed after regular exercise.
Patients should be advised to pay attention to possible symptoms of coronary heart disease, even if they are trained exercisers.
Hypertrophic cardiomyopathy, anomalous coronary artery of wrong sinus origin, myocarditis, and arrhythmogenic right ventricular cardiomyopathy [18, 19] are an exception to the lower overall risk associated with intensive exercise that occurs in patients with certain, often unrecognized underlying heart diseases.
A family history of SCA, which could be associated with myocardial infarction, is related to a 1.5- to 1.8-fold increased risk of SCA [20]. Traditional risk factors that tend to aggregate in families, such as hypercholesterolemia, hypertension, diabetes mellitus, and obesity, do not explain the increase in risk. However, it is likely that interactions of mutations or polymorphisms in specific genes and environmental factors influence this risk.
Elevated serum C-reactive protein (CRP) is also associated with an increased risk of SCA [21]. Higher serum concentrations of CRP could be an expression of chronic inflammation that has been implicated as a risk factor for cardiovascular diseases (including acute coronary syndromes and stroke).
Heavy alcohol consumption (six or more drinks per day) or binge drinking increases the risk for SCD [22, 23]. In fact, moderate alcohol intake (e.g., one to two drinks per day) is related to a reduction of the risk of SCD [22].
Clinical observations have suggested that acutely stressful situations could have a possible relation with a higher risk of SCA. For example, major disasters, such as earthquakes and war, result in a rapid transient increase in the rate of SCA in populations [15]. Social support from others may reduce the risk associated with stressful life events.
Excessive caffeine intake has been investigated as a potential risk factor for SCA [24], but no significant association with SCA has been found.
After a myocardial infarction, elevated plasma nonesterified fatty acid (free fatty acid) concentrations were associated with ventricular arrhythmias and SCD [25]. However, in the Cardiovascular Health Study, in a population-based cohort of older adults [26], nonesterified fatty acids were not associated with SCD. Moreover, in a population-based case-control study among people without prior clinically recognized heart disease, cases of SCA had higher concentrations of trans isomers of linoleic acid in red blood cell membranes [27].
In contrast, in several studies, a higher dietary intake and higher plasma levels of long-chain n-3 polyunsaturated fatty acids (eicosapentaenoic acid and docosahexaenoic acid) are associated with a lower risk of SCD [28].
Mechanisms of Sudden Cardiac Death
The rhythm most often recorded at the time of sudden cardiac arrest is VF. Previous studies suggest that up to 80 % occur via this mechanism and up to 20 % are attributed to bradyarrhythmias, including advanced atrioventricular (AV) block and asystole [1]. Bayes de Luna et al. [31] reported that in 157 ambulatory patients who had SCD while undergoing Holter recording, 62.4 % had VF, 16.5 % had bradyarrhythmias, 12.7 % had torsades de pointes, and 8.3 % had primary VT. The true incidence of bradyarrhythmias is not easy to understand since a rhythm beginning as VF may appear to be asystole during the first ECG recording. Advanced AV block or significant bradycardia can provoke VF. It is difficult to identify clearly the electrophysiological mechanism responsible for SCD. Mechanisms may be multifactorial and are different depending on the specific cardiac abnormality, and a rhythm can begin via one mechanism and proceed via a different one. Class I and III antiarrhythmic agents were not able to reduce total and SCD mortality in patients at risk for SCD [30]. In fact, both classes do not direct their actions on cardiac muscle or specialized conducting tissues that have been shown effective for prevention of SCD. Beta-blockers, ACE inhibitors, angiotensin receptor-blocking agents, lipid-lowering agents, spironolactone, and fibrinolytic and antithrombotic agents were judged likely effective especially in primary prevention. Since SCD is for the most part the result of a ventricular tachyarrhythmia, these drugs act on the biochemical, ischemic, and fibrotic processes that underlie the onset or maintenance of life-threatening ventricular arrhythmias.
The mechanisms of cardiac arrest are represented by electromechanical dissociation, asystole and heart block, and VF. More permanent changes could also occur, such as plaque rupture, but it is likely that transient factors interact with a fixed substrate to precipitate the arrhythmia. The possibilities include physical activity, transient ischemia, hypoxia, stretch, ion channel abnormalities, pH and electrolyte changes, inflammation, neuroendocrine actions, and drugs.
Management
The acute management of survivors of SCA includes the following:
1.
Identification and treatment of acute reversible causes
2.
Evaluation for structural heart disease
3.
Evaluation for primary electrical diseases in patients without identifiable arrhythmic triggers or cardiac structural abnormalities
4.
Neurologic and psychologic assessment
5.
Evaluation of family members in selected patients with a suspected or confirmed heritable syndrome
Noninvasive Management
ECG
A noninvasive evaluation is recommended with resting 12-lead ECG in all patients who are evaluated for ventricular arrhythmias (class I, level of evidence: A). It allows to identify various congenital abnormalities associated with ventricular arrhythmias and SCD (e.g., LQTS, SQTS, Brugada syndrome, ARVC) but also other ECG modifications, such as those due to electrolyte disturbances, or evidence suggesting underlying structural disease, such as bundle branch block; Q waves indicative of ischemic heart disease, ventricular hypertrophy, or infiltrative cardiomyopathy; and AV block. QRS duration and repolarization abnormalities are both independent predictors of SCD. A QRS duration greater than 130 ms has been associated with increased mortality in patients with a reduced LVEF (equal to or less than 30 %). Prospective studies have also reported an association between ST-segment depression or T-wave abnormalities and increased risk of cardiovascular death and SCD in particular [1]. QTc greater than 440 ms has been demonstrated to predict cardiovascular death with adjusted relative risk of 2.1 [31]. Some data suggest that the correlation between QTc and survival could be “J shaped.” So, also relatively short QTc intervals have also been associated with increased risk. The definition of short QT syndrome as a QTc less than 300 ms has been proven to be an independent predictor of SCD [32].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


