Fig. 1.1
(a, b) Patient ECG: sinus rhythm, third-grade atrioventricular block, narrow QRS escape rhythm with a heart rate of 33 bpm, ST-segment elevation in the inferior leads (DII, DIII, aVf) with specular ST-segment depression in DI and aVl, QRS axis of 105°
A complete echocardiographic examination was performed and showed normal dimensions of the cardiac chambers, reduced systolic left ventricle function (EF 45 %) due to hypokinesia of the inferior and posterolateral wall, normal right ventricular function (TAPSE 21 mm), and mild mitral and tricuspid regurgitation with normal pulmonary artery systolic pressure.
Clinical Course and Therapeutic Management
These findings all together were suggestive for inferior ST elevation acute coronary syndrome complicated by third-degree AV block.
As soon as possible, a transcutaneous pacing and a continuous electrocardiographic, blood pressure, and oxygen saturation monitoring were placed, and the cath lab team was advised. Therapy with crystalloid and dopamine 5 gamma/kg/min infusion was practiced with blood pressure increase to 120/70 mmHg, and morphine was administered for the transcutaneous pacing pain. At 1:45 AM the patient was transferred to the cath lab to perform coronary angiography and to position a temporary pacing via the right femoral vein. The angiography showed right coronary artery dominance with thrombotic occlusion of its middle tract; no other stenosis was present. At 2:05 AM the patient underwent manual thrombus aspiration (EXPORT) and PTCA with medicated self-expanding stent (STENTYS DES 3.5−4.5 × 17 mm) implantation via the right radial artery. A bolus of abciximab 0.25 mg/kg was administered, and the patient was shifted to prasugrel with a loading dose of 60 mg administered in the cath lab. The residual stenosis was <20 % and TIMI flow was 3. Continuous unfractionated heparin infusion was administered during the 12 h following the procedure. The transvenous pacing was removed 48 h later being the patient again in stable sinus rhythm. Continuous electrocardiographic monitoring during the first 6 days did not show any dangerous tachyarrhythmia or bradyarrhythmia. Therapy with ASA 100 mg od, prasugrel 10 mg od, atorvastatin 80 mg od, losartan 12.5 mg od, and pantoprazole 40 mg od was started since the first day. Metoprolol tartrate 25 mg bid was added the second day. The patient was transferred to our semi-intensive cardiology unit on the third day and then discharged on the seventh day with a follow-up visit, ECG, and echocardiography programmed 2 months later. Therapy at discharge was ASA 100 mg od, prasugrel 10 mg od for 12 months, atorvastatin 80 mg od, losartan 12.5 mg od, pantoprazole 40 mg, and metoprolol tartrate 25 mg bid.
1.2 ST Elevation Myocardial Infarction (STEMI)
Definition and Epidemiology
The last ESC guidelines published in 2012 define “acute myocardial infarction” (AMI) as the evidence of myocardial necrosis (elevation of cardiac biomarkers, typical ECG alterations, imaging alterations, or autopsy evidence) in the presence of a clinical setting suggestive for myocardial ischemia [1].
Acute myocardial infarction with ST elevation (STEMI) is a clinical syndrome characterized by the typical symptoms of myocardial ischemia with electrocardiographic ST elevation (persistent for more than 20 min) and following release of cardiac biomarkers [2].
Coronary artery disease (CAD) is the most common cause of death in the whole world. In 2012, 7.4 million people in Europe (which is 13.2 % of all deaths) died from CAD [3].
At present, up to 25–40 % of AMI presentations are STEMI ones [4–7]. The incidence of STEMI hospital admissions is different among countries that belong to ESC [8]. In the last decades there has been a STEMI incidence decrease despite an NSTEMI incidence increase. In the Sweden registry, which is probably the most comprehensive STEMI registry, an incidence of 66 STEMI/100,000/year, similar to those of other countries like the Czech Republic [9], Belgium [8], and the USA [10], has been reported.
The in-hospital mortality of unselected STEMI patients varies from 6 to 14 % [8]. Ejection fraction, the Killip class, age, time delay to treatment and mode of treatment, prior myocardial infarction history, renal dysfunction, diabetes mellitus, and diseased coronary artery number are all factors that influence mortality. Hospitals with a high clinical volume and high rate of invasive procedures have lower mortality rates [11]. STEMI mortality significantly decreased thanks to a frequency care increase [5, 7].
Pathology and Pathophysiology
Pathology
The causes of STEMI are different, but they can be divided into two principal groups:
Coronary atherosclerosis complicated by coronary thrombosis, the main one
Non-atherogenic forms that are rare such as arteritis, trauma to coronary arteries, coronary mural thickening with metabolic disease or intimal proliferative disease, emboli to coronary arteries, congenital coronary artery anomalies, myocardial oxygen demand–supply disproportion, hematologic, and miscellaneous [12]
We will focus our attention on the first cause of STEMI, the coronary atherosclerosis.
A previous classification based on ECG evolution divided patients with MI into two groups: patients with a Q-wave infarction, very often considered a transmural infarction, and patients with a non-Q-wave infarction. Currently, a new classification based on pathophysiology divides patients into other two groups: those with STEMI related to an acute thrombotic occlusion of an epicardial coronary artery and those with NSTEMI/unstable angina, due to stenosis of a coronary artery without occlusive thrombi. When there is a chronic total occlusion of the coronary artery, patients do not always have an MI because of collateral blood flow development and other factors.
The most important element on AMI’s physiopathology is the atherosclerotic plaque. The plaque evolution is an active process lasting years that consists in intima lipoprotein accumulation, lipoprotein oxidation and glycation, intima leukocyte migration, foam cell development, intima smooth cell migration with consequent extracellular matrix accumulation, atherosclerotic plaque growth, and a fibrofatty lesion formation with a lipid core surrounded by an acellular fibrous capsule. Cytokines and effector molecules like hypochlorous acid and superoxide anion play an important role in this process.
During this natural evolution, high-risk plaques can undergo plaque disruption [13, 14] that is induced by stressors like intramural blood pressure, coronary vasomotor tone, and tachycardia. So, thrombogenic substances are exposed with secondary activation and aggregation of platelet; moreover, thrombin generation is promoted with subsequent thrombus formation. There is seasonal and circadian variation of some of these key physiologic variations, and that is why STEMI happens more frequently in the winter early morning hours and following natural disasters [15].
Anatomically, two major types of MI can be detected: transmural infarcts characterized by the presence of a full ventricular wall thickness myocardial necrosis and nontransmural or subendocardial MI with necrosis involvement of the subendocardium or intramural myocardium or both. In the first case, there is a completely occlusive thrombus of an epicardial coronary artery that subtends the infarct area with a typical ST-segment elevation. The transmural necrosis can cause a full wall thickness vital myocardial loss and subsequent fibrosis that is evidenced by Q-wave evolution in the leads overlying the infarcted zone. In a few number of patients, there is not a Q-wave evolution but an R-wave height reduction.
There is a specific correlation between the coronary artery occluded, the myocardial area developing necrosis, and the ECG derivation that shows an ST elevation (Fig. 1.2).
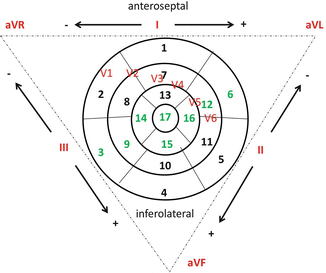
Fig. 1.2
Schematic representation of left ventricle segments, Einthoven’s triangle with electrocardiogram derivations. LAD supplies segments nr 1, 2, 6, 7, 8, 12, 13, 14, 15, 16, and 17; LCX supplies segments nr 4, 5, 6, 10, 11, 12, 15, 16, and 17; RCA supplies segments nr 3, 4, 9, 10, 11, 14, 15, 16, and 17. Areas of shared perfusion between LAD, LCX, and RCA are shown in green. The infarct artery can be deduced identifying the leads with ST-segment elevation and correlating these with the segments that these leads explore and so with coronary arteries that supply these segments. For example, ST segment elevated prominently in leads exploring segments 1, 2, 7, 8, 13, 14, and 17 which means that the occluded vessel is the LAD. LAD, left anterior descending; RCA, right coronary artery; LCX, left circumflex
The earliest myocardial ultrastructural changes occurring within the first 20 min are reversible. Changes become irreversible after 20 min up to 120 min of ischemia [16]. After 6–12 h of necrosis onset, myocardium gross alteration can be identified. So, in the first 30 min after ischemia onset, myocardial injury is reversible; subsequently, a progressive viability loss occurs and usually completes at 6–12 h. That is the reason why the reperfusion therapy benefits are greatest when patients are treated early.
Generally, the right ventricle is less involved by infarction. It is interested in approximately 50 % of patients with transmural infarct of the inferoposterior wall and posterior portion of the septum [17]. The right ventricle shows an excellent recovery of systolic function once reperfusion is restored [18].
Pathophysiology
When a coronary artery is occluded, immediate myocardial contractile alteration occurs. There are four sequential patterns of abnormal contraction:
Dyssynchrony (adjacent segments do not contract at the same time)
Hypokinesia (reduced contraction)
Akinesia (cessation of contraction)
Dyskinesia (the segment expansion is paradoxical)
As an acute compensation of these alterations, a hyperkinesia of the normal myocardium segments usually firstly develops. It results from sympathetic nervous system activity increase and Frank–Starling mechanism and lasts up to 2 weeks.
If ischemic injury involves >15 % of the myocardium, the systolic–diastolic function of the LV becomes depressed, and a decline of cardiac output, stroke volume, and blood pressure occurs. End-systolic volume and end-diastolic pressure increase and diastolic dysfunction appears. The degree of end-systolic volume increase has been shown to be an important predictor of STEMI mortality [19]. If ischemic injury involves >25 % of the myocardium, clinical heart failure becomes typical, and if the myocardial loss is >40 %, cardiogenic shock appears.
Improvement is possible thanks to the recovery of the stunned (reversibly injured) myocardium after revascularization, but in some patients the infarcted LV may dilate causing LV remodeling. This can be a compensatory mechanism restoring a normal stroke at the expense of a reduced ejection fraction; however, dilation elevates afterload (Laplace’s law) that depresses LV stroke volume and increments the consumption of myocardial oxygen, intensifying myocardial ischemia. The infarct size, patency of the related coronary artery, and renin–angiotensin–aldosterone system (RAAS) influence LV remodeling. For this reason, LV remodeling can be reduced by an antagonist of RAAS. Even aldosterone inhibitors reduce collagen deposition and decrease ventricular arrhythmia development [20].
Clinical Features
Symptoms
The typical STEMI discomfort is a prolonged (more than 20 min), constricting, oppressing, or compressing pain of variable intensity. It has a retrosternal location and often radiates to the ulnar side of the left arm, or rarely both arms; to the neck, jaw, and shoulders; and rarely to the epigastrium or interscapular region. In some patients, frequently those with an inferior STEMI, the location is the epigastrium simulating abdominal disorders. In these patients nausea and vomiting may occur due to vagal reflex activation or LV receptor stimulation. Symptoms like cold perspiration, palpitations, profound weakness, dizziness, and a sense of imminent death may be present.
In some cases there is an atypical presentation of STEMI-like atypical location of the pain or dyspnea, syncope, profound weakness, or acute indigestion. Some patients are wholly asymptomatic, and STEMI can be unrecognized and discovered in a subsequent routine electrocardiographic examination. These patients have a similar prognosis of symptomatic ones.
In up to half of STEMI patients, a precipitating factor like reduced oxygen supply to the myocardium (hypotension, hypoxemia, pulmonary embolism, etc.) or increased myocardial oxygen demands (aortic stenosis, fever, agitation, tachycardia, emotional stress, unusually heavy exercise) can be identified.
Physical Examination
STEMI patients may appear anxious and agitated. Heart rate varies from bradycardia to tachycardia/tachyarrhythmia, and blood pressure varies from hypotension (patient with right ventricle involvement or cardiogenic shock or low blood pressure acute heart failure) to normotension and even to hypertension due to adrenergic activation.
Fever is present in most patients and resolves within 4–5 days. It is a nonspecific response to tissue necrosis.
Patients with cardiogenic shock or right ventricular infarction infarct have elevated jugular pressure.
Carotid pulse in STEMI patients may be small due to reduced stroke volume. When LV failure develops, rales are audible.
The Killip classification is a prognostic classification dividing STEMI patients according to the presence and severity of heart failure signs:
Class I: no rales or third sound
Class II: rales in <50 % of pulmonary field with or without third sound
Class III: rales in >50 % of pulmonary field (pulmonary edema)
Class IV: cardiogenic shock
A third and/or fourth sound may be heard in STEMI patients with severe LV dysfunction that determines elevation of LV filling pressure. When the fourth sound is heard, a corresponding presystolic pulsation is present. Additional systolic murmur (transient/persistent) due to mitral regurgitation as a result of mitral valve apparatus dysfunction may be audible. Along the left and right sternal border, a holosystolic, prominent murmur, accompanied by a thrill, is audible in the presence of interventricular septum rupture. Pericardial friction rubs can be present especially in patients with large infarctions [21]. They are audible along the left sternal border in the first 2 weeks and most commonly on the second or third day.
Diagnosis
The diagnosis of STEMI starts from symptom assessment: history of chest pain lasting at least 20 min or more; not responding to nitroglycerine is typical.
The confirmation of diagnosis must be completed as soon as possible with a 12-lead ECG, also considering the addition of posterior (V7–V8–V9) or right leads (V4R–V5R, V6R) in patients with high suspicion, respectively, of posterior or right ventricle infarction.
If available, a continuous ECG monitoring should be initiated in all patients to detect life-threatening arrhythmias and allow defibrillation if required.
The diagnostic electrographic sign is a new ST-segment elevation measured at the J point in two contiguous leads with the following thresholds: ≥0.25 mV in men below the age of 40 years, ≥0.2 mV in men over the age of 40 years, or ≥0.15 mV in women in leads V2–V3 and/or ≥0.1 mV in other leads [1]. According to leads involved by ST elevation, the localization of the ischemia is as follows:
Anterior MI: V1–V6
Septal MI: V1–V4
Lateral MI: I, aVL, V5, V6
Inferior MI: II, III, aVF
Posterior MI: V7–V8–V9 (high R in V1–V3 with ST depression V1–V3)
Right ventricle MI: V1, V4R–V5R–V6R
Although not frequently seen, an earlier sign of ischemia could be the presence of hyperacute T waves; later, the ECG alterations evolve in ST elevation in those leads that register the electrical activity of the ischemic myocardium. ST elevation typically presents a concave configuration but over time becomes more pronounced, more convex, and rounded upward. In the absence of reperfusion strategies, the natural evolution of ECG is as follows: the ST gradually returns to isoelectric baseline, there is a reduction of R-wave amplitude with the development of Q waves, and T waves become inverted. The ECG changes usually may take place from few weeks to several hours from presentation.
Moreover, the initial ECG presentation of acute coronary syndrome could be represented by new or presumed new left bundle branch block (LBBB) [1].
The electrocardiographic diagnosis could be more difficult in some categories of patients:
Patients with preexistent LBBB: in the presence of intraventricular conduction delay, the diagnosis could be suspected in the presence of concordant ST elevation with QRS or in case of marked ST abnormalities. Two signs are highly specific: Cabrera’s sign, a prominent notching in the ascending limb of S wave in leads V3–V4, and Chapman’s sign, a notching in the ascending limb of R wave in V5–V6 [22]. A scoring system has been developed from the GUSTO-1 trial called Sgarbossa’s criteria [23] but not providing diagnostic certainty.
Patients with paced rhythm: in case of clinical strong suspicion, the diagnosis should be confirmed by angiography; reprogramming of the pacemaker with the appearance of intrinsic rhythm and the evaluation of ischemic ECG changes may be considered when feasible.
Patients with isolated posterior myocardial infarction: the involvement of the inferobasal portion of the heart may appear as an isolated ST depression ≥0.05 mV in leads V1 through V3. The documentation of ST elevation ≥0.05 mV in the posterior chest wall leads should be treated as STEMI.
Patients with left main coronary obstruction: the typical ECG signs are aVR ST elevation and inferolateral ST depression; the presence of ST depression in eight or more surface leads together with ST elevation in aVR and/or V1 suggests ischemia due to multivessel disease or left main coronary artery obstruction.
The following steps are not necessary for the diagnosis; however, they complete the clinical picture of patients with ACS: blood sampling for troponin determination and echocardiography for differential diagnosis and for the assessment of the involved myocardium, left ventricular function, and mechanical complications.
Therapy
The following recommendations are based on currently accepted European guidelines [1].
Initial therapy of patients with acute coronary syndrome with ST elevation is represented as follows:
Oxygen administration in the presence of hypoxia or acute heart failure.
Relief of pain and anxiety: IV opioids are very useful, although they must be used with caution for their potential side effects, such as respiratory depression, nausea, vomiting, hypotension, and bradycardia.
The following steps of treatment are related to some crucial aspects: the first one is time from symptom onset and the second one, the availability of a primary PCI center.
Patients with a diagnosis of STEMI within 12 h from symptom onset should be considered for mechanical or pharmacological reperfusion strategy, as soon as possible. Moreover, reperfusion therapy should be taken into account in the presence of ongoing ischemia even if the onset of pain dates back more than 12 h. Primary PCI may also be done in stable patients presenting 12–24 h after symptom onset.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


