Benjamin M. Scirica, David A. Morrow
ST-Elevation Myocardial Infarction
Pathology, Pathophysiology, and Clinical Features
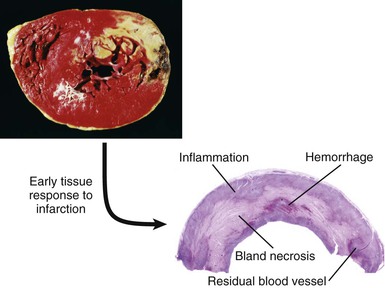
Pathologic diagnosis of myocardial infarction (MI) requires evidence of myocardial cell death caused by ischemia. Characteristic findings include coagulation necrosis and contraction band necrosis, often with patchy areas of myocytolysis at the periphery of the infarct. During the acute phase of MI, myocytes die in the infarct zone, with subsequent inflammation, clearance of necrotic debris, and repair eventuating in scar formation.
Clinical diagnosis of MI requires a clinical syndrome indicative of myocardial ischemia with some combination of evidence of myocardial necrosis on biochemical, electrocardiographic, or imaging modalities. The sensitivity and specificity of the clinical tools for diagnosing MI vary considerably depending on the timing of evaluation after the onset of infarction. Cardiac professional societies have jointly established updated criteria for the diagnosis of MI (Table 51-1).1 The revised universal definition of myocardial infarction classifies MI into five types, depending on the circumstances in which the MI occurs (Table 51-2).1 These revisions to the definition of MI and a shift to more sensitive biomarkers of myocardial injury have important implications not only for the clinical care of patients but also for epidemiologic study, public policy, and clinical trials.2,3
TABLE 51-1
Universal Definition of Myocardial Infarction

CABG = coronary artery bypass grafting; cTn = cardiac troponin; LBBB = left bundle branch block; URL = upper reference limit.
From Thygesen K, Alpert JS, White HD, et al: Universal definition of myocardial infarction. J Am Coll Cardiol 60:1581, 2012.
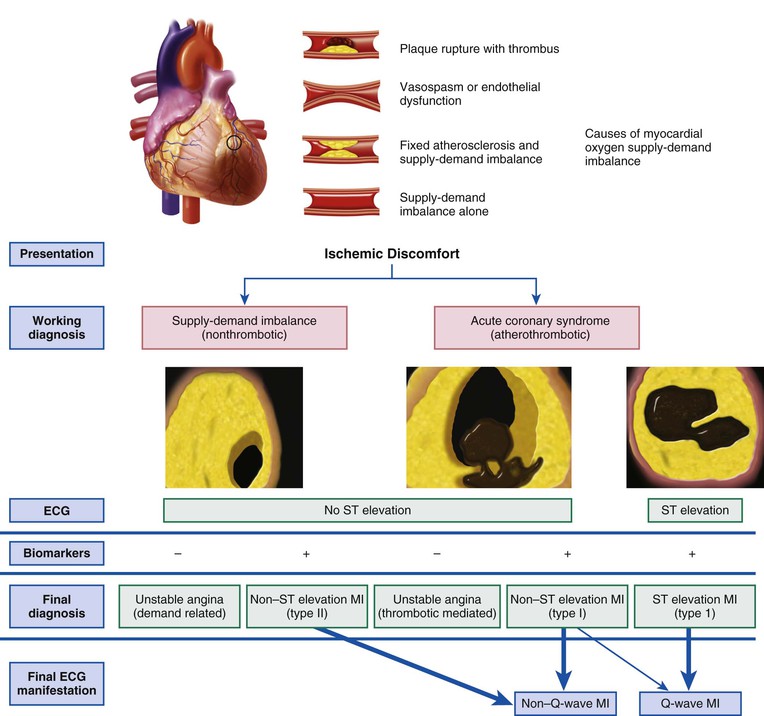
The contemporary approach to patients with ischemic discomfort is to consider them to be experiencing an acute coronary syndrome (ACS), which encompasses the diagnoses of unstable angina, non–ST-segment elevation MI (NSTEMI), and ST-segment elevation MI (STEMI) (Fig. 51-1). The principal diagnostic tool for patients with suspected ACS is the 12-lead electrocardiogram (ECG), which discriminates those with ST-segment elevation, the subject of Chapters 51 and 52, and those without ST-segment elevation, the subject of Chapter 53.
Changing Patterns in Incidence and Care
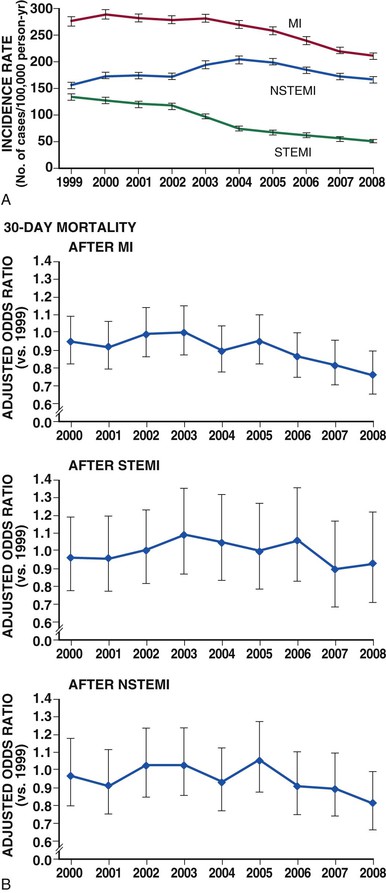
Despite advances in diagnosis and management, STEMI remains a major public health problem in the industrialized world and is on the rise in developing countries (see Chapter 1).4 In the United States, almost 600,000 patients are admitted to the hospital each year with a primary diagnosis of ACS. The number exceeds 1 million with the inclusion of ACS as a secondary diagnosis.5 The rate of MI rises sharply in both men and women with increasing age, and racial differences exist, with MI occurring more frequently in black men and women regardless of age. The proportion of patients with ACS events who have STEMI varies across observational studies—from 29% to 47% of patients admitted with ACS. This estimate does not include “silent” MI, which may not prompt hospitalization. Between 1999 and 2008, the proportion of patients with an ACS and STEMI declined by almost 50% (Fig. 51-2A; see also Fig. 53-2).6
Of particular concern from a global perspective, the burden of MI in developing countries may be approaching that now afflicting developed countries.4 The limited resources available to treat STEMI in developing countries mandate major international efforts to strengthen primary prevention programs (see also Chapter 1).
Improvements in Outcome
The overall number of deaths from STEMI has declined steadily over the past 30 years, but it has stabilized over the past decade (Fig. 51-2B).6–9 Both a decreased incidence of STEMI and a decline in the case fatality rate after STEMI have contributed to this trend.5 According to estimates from the American Heart Association, the short-term mortality rate of patients with STEMI ranges from 5% to 6% during the initial hospitalization and from 7% to 18% at 1 year.10 Mortality rates in clinical trial populations tend to be approximately half of those observed in registries of consecutive patients, most likely because of the exclusion of patients with more extensive comorbid medical conditions.
Improvements in the management of patients with STEMI have occurred in several phases.11 The “clinical observation phase” of coronary care consumed the first half of the 20th century and focused on detailed recording of physical and laboratory findings, with little active treatment of the infarction. The “coronary care unit phase” began in the mid-1960s and emphasized early detection and management of cardiac arrhythmias based on the development of monitoring and cardioversion/defibrillation capabilities. The “high-technology phase,” heralded by the introduction of the pulmonary artery balloon flotation catheter, set the stage for bedside hemodynamic monitoring and directed hemodynamic management. The modern “reperfusion era” of STEMI care began with intracoronary and then intravenous fibrinolysis, increased use of aspirin (see Chapter 52), and subsequently the development of primary percutaneous coronary intervention (PCI) (see Chapter 55).
Contemporary care of patients with STEMI has entered an “evidence-based coronary care phase,” which is increasingly being influenced by guidelines and performance measures for clinical practice.10,12,13 Implementation of guideline-directed medical treatment (GDMT) and regional quality initiatives has significantly decreased heterogeneity in care, increased compliance with evidence-based therapies, and improved outcomes.14,15 Mandatory outcome and procedural reporting has resulted in the establishment of benchmarks for procedural success and mortality rates in patients with MI cared for at various hospitals (www.hospitalcompare.hhs.gov).
Limitations of Current Therapy
Rates of appropriate initiation of reperfusion therapy vary widely, with up to 30% of patients with STEMI who are eligible to receive reperfusion therapy not receiving this lifesaving treatment in some registries.16 Therefore initiatives to increase timely administration of guideline-directed reperfusion therapy are important to achieve improvements in care (see Chapter 52).
Advanced age is a principal determinant of mortality in patients with STEMI.17,18 Cardiac catheterization and other invasive procedures are being performed more commonly during hospitalization in elderly patients with STEMI. Nevertheless, evidence suggests that the greatest reductions in mortality in elderly patients are gained by strategies used during the first 24 hours, a time frame in which prompt and appropriate use of lifesaving reperfusion therapy is paramount—thus emphasizing the need to extend advances in GDMT for STEMI to older adults.19
Management and outcomes of patients with STEMI appear to vary substantially depending on the volume of such patients cared for within a hospital system.20,21 Mortality rates in patients with STEMI are lower in hospitals with a high clinical volume, a high rate of invasive procedures, and a top ranking in quality reports. Conversely, patients with STEMI not cared for by a cardiovascular specialist have higher mortality rates. Variation also occurs in the treatment patterns of certain population subgroups with STEMI, notably women and blacks, although after adjusting for comorbid conditions and the degree of atherosclerosis, outcomes appear to be similar.22
Pathologic Findings
Based on research beginning in the 1970s, we now recognize that almost all ACS events result from coronary atherosclerosis, generally with superimposed coronary thrombosis caused by rupture or erosion of an atherosclerotic lesion.23,24 Nonatherogenic forms of coronary artery disease are discussed later in this chapter, and causes of MI without coronary atherosclerosis are presented in Table 51-3.
TABLE 51-3
Causes of Myocardial Infarction Without Coronary Atherosclerosis
| Coronary Artery Disease Other than Atherosclerosis |
Granulomatous (Takayasu disease) Mucocutaneous lymph node (Kawasaki) syndrome Disseminated lupus erythematosus Radiation (radiation therapy for neoplasia) Coronary mural thickening with metabolic disease or intimal proliferative disease Mucopolysaccharidoses (Hurler disease) Juvenile intimal sclerosis (idiopathic arterial calcification of infancy) Intimal hyperplasia associated with contraceptive steroids or with the postpartum period Coronary fibrosis caused by radiation therapy Luminal narrowing by other mechanisms Spasm of coronary arteries (Prinzmetal angina with normal coronary arteries) Spasm after nitroglycerin withdrawal |
| Emboli to Coronary Arteries |
| Congenital Coronary Artery Anomalies |
| Myocardial Oxygen Demand-Supply Disproportion |
| Hematologic (In Situ Thrombosis) |
| Miscellaneous |

Modified from Cheitlin MD, McAllister HA, de Castro CM: Myocardial infarction without atherosclerosis. JAMA 231:951, 1975. Copyright 1975, American Medical Association.
When acute coronary atherothrombosis occurs, the resulting intracoronary thrombus may be partially obstructive, which generally leads to myocardial ischemia in the absence of ST-segment elevation, or be completely occlusive and cause transmural myocardial ischemia and STEMI. Before the fibrinolytic era, clinicians typically divided patients with MI into those in whom a Q wave developed on the ECG and those with non–Q-wave MI based on evolution of the ECG pattern over several days. The term Q-wave infarction was frequently considered to be virtually synonymous with transmural infarction, whereas non–Q-wave infarctions were often referred to as subendocardial infarctions. Contemporary studies using cardiac magnetic resonance (CMR) indicate that the development of a Q wave on the ECG is determined more by the size of the infarct than by the depth of mural involvement. Thus use of ACS as the more appropriate broad conceptual framework has replaced this terminology, anchored by the underlying unifying pathophysiology (see Fig. 51-1). Further classification of patients by the presence of ST-segment elevation (STEMI) or by its absence (non–ST-segment elevation ACS) rather than by the evolution of Q waves is preferable because immediate clinical decisions such as fibrinolysis or primary PCI depend on identification of diagnostic ST-segment elevation on the initial ECG.
Plaque (See also Chapter 41)
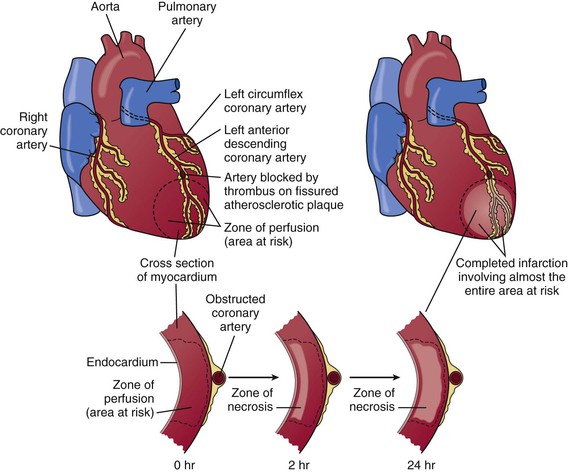
Atherosclerotic plaque begins early in life and grows slowly over decades.25 Evidence of some atherosclerosis is almost ubiquitous in the modern world, yet most plaque remains asymptomatic throughout a lifetime. Other plaques may develop slowly and elicit stable symptoms. Plaque that precipitates an ACS event through the abrupt and catastrophic transition from a vulnerable, yet stable plaque to an unstable one characterized by plaque disruption or erosion and then subsequent overlying thrombosis is rare.23,26 Traditional risk factors and consequent chronic inflammation promote much of the development of atherosclerosis, although some patients have a systemic predisposition to plaque disruption that is independent of traditional risk factors. Plaque disruption exposes substances that promote platelet activation and aggregation, thrombin generation, and ultimately thrombus formation.23,26 The resultant thrombus interrupts blood flow and leads to an imbalance between oxygen supply and demand and, if this imbalance is severe and persistent, to myocardial necrosis (Fig. 51-3).
Composition of Plaque
The atherosclerotic plaque associated with total thrombotic occlusion of an epicardial coronary artery, located in infarct-related vessels, is generally more complex and irregular than the plaque in vessels not associated with STEMI.23,26 Histologic studies of these lesions often reveal plaque rupture or erosion (see Chapter 41). Thrombus composition may vary at different levels: white thrombi contain platelets, fibrin, or both, and red thrombi contain erythrocytes, fibrin, platelets, and leukocytes (see Chapter 82).
Plaque Fissuring and Disruption
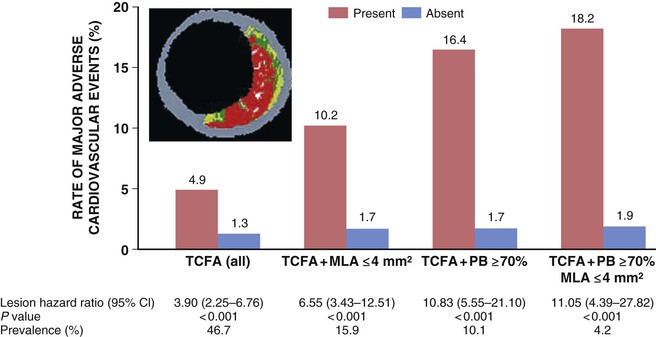
In autopsy studies, plaque rupture and plaque erosion are the most common underlying causes of MI and sudden cardiac death. Plaque rupture is present in almost three quarters of cases and is more prevalent in men. Plaque erosion is more frequent in women younger than 50 years, although the prevalence of rupture increases as women age.23 Atherosclerotic plaque considered prone to disruption or erosion is most likely plaque that has evolved to a morphology that includes a necrotic core filled with lipids and inflammatory cells and covered by a thin and inflamed fibrous cap. A prospective study of 697 patients with ACS who underwent three-vessel coronary angiography and gray-scale radiofrequency intravascular ultrasonographic imaging after PCI found that three lesion characteristics—lipid burden greater than 70%, thin-cap fibroatheroma morphology, and a minimal luminal area of 4.0 mm2 or smaller—were independent correlates of future atherosclerotic events (Fig. 51-4).27 Other morphologic characteristics associated with rupture-prone plaque include expansive remodeling that minimizes luminal obstruction (mild stenosis by angiography), neovascularization (angiogenesis), plaque hemorrhage, adventitial inflammation, and a “spotty” pattern of calcification.23
Inflammation stimulates the overexpression of enzymes that degrade components of the plaque’s extracellular matrix.23,26 Activated macrophages and mast cells, abundant at the site of plaque disruption in patients who die of STEMI, can elaborate these proteinases. In addition to these structural aspects of vulnerable or high-risk plaque, stress induced by intraluminal pressure, coronary vasomotor tone, tachycardia (cyclical stretching and compression), and disruption of microvessels combines to produce plaque disruption at the margin of the fibrous cap near an adjacent, less involved segment of the coronary artery wall (shoulder region of the plaque).25 Several key physiologic variables—such as systolic blood pressure, heart rate, blood viscosity, endogenous tissue plasminogen activator (t-PA) activity, plasminogen activator inhibitor-1 (PAI-1) levels, plasma cortisol levels, and plasma epinephrine levels—exhibit circadian and seasonal variations and increase at times of stress. These factors act in concert to heighten the propensity for plaque disruption and coronary thrombosis, with the result that STEMI clusters in the early morning hours, especially in the winter and after natural disasters.28–30
Acute Coronary Syndromes
Plaque disruption exposes thrombogenic substances that may produce an extensive thrombus in the infarct-related artery (see Fig. 51-1). An adequate collateral network that prevents necrosis from occurring can result in clinically silent episodes of coronary occlusion; in addition, many plaque ruptures are asymptomatic if the thrombosis is not occlusive. Characteristically, completely occlusive thrombi lead to transmural injury to the ventricular wall in the myocardial bed subtended by the affected coronary artery (Fig. 51-5; see also Figs. 51-1 and 51-3). Infarction alters the sequence of depolarization ultimately reflected as changes in the QRS.31 The most characteristic change in the QRS that develops in most patients with STEMI is the evolution of Q waves in leads overlying the infarct zone (see Figs. 51-1 and 51-5).31 In a minority of patients with ST elevation, no Q waves develop but other abnormalities in the QRS complex occur frequently, such as diminution in R wave height and notching or splintering of the QRS (see Chapter 12). Patients who have ischemic symptoms without ST elevation are initially diagnosed as suffering either from unstable angina or, with evidence of myocardial necrosis, from NSTEMI (see Fig. 51-1).
Patients with persistent ST-segment elevation are candidates for reperfusion therapy (either pharmacologic or catheter based) to restore flow in the occluded epicardial infarct-related artery (see Chapter 52).10 ACS patients without ST-segment elevation are not candidates for pharmacologic reperfusion but should receive anti-ischemic therapy, followed in most cases by PCI (see Chapter 53). Thus the 12-lead ECG remains at the center of the decision pathway for the management of patients with ACS to distinguish between those with ST elevation and those without it (see Figs. 51-1 and Fig. 55-5).32
Heart Muscle
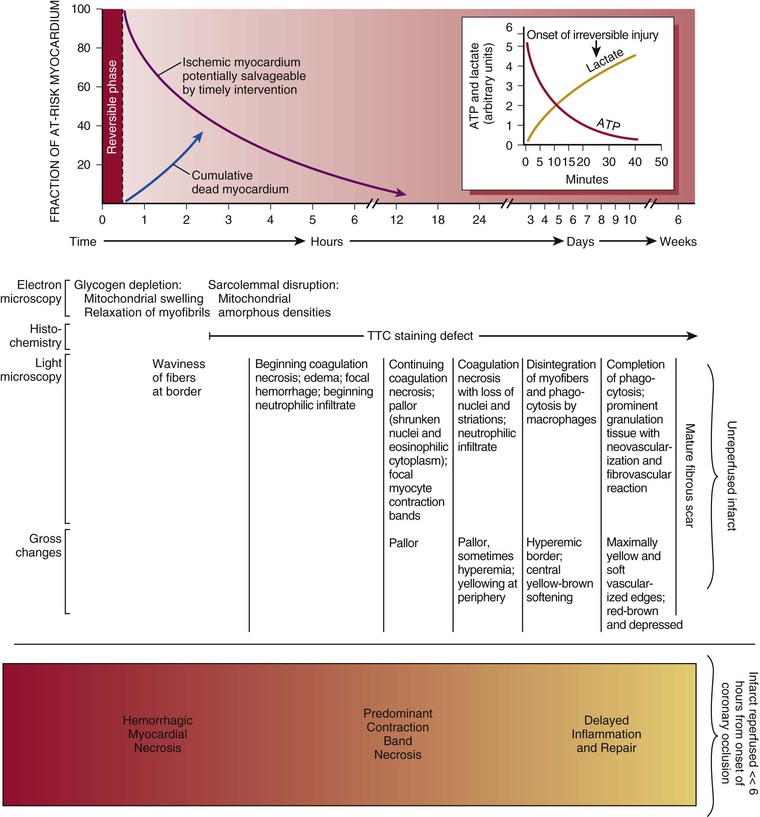
The cellular effects of ischemia commence within seconds of the onset of hypoxia with the loss of adenosine triphosphate (ATP) production. Myocardial relaxation-contraction is compromised, and irreversible cell injury begins within as early as 20 minutes. Necrosis is usually complete in 6 hours unless reperfusion occurs or an extensive collateral circulation is present (Fig. 51-6).
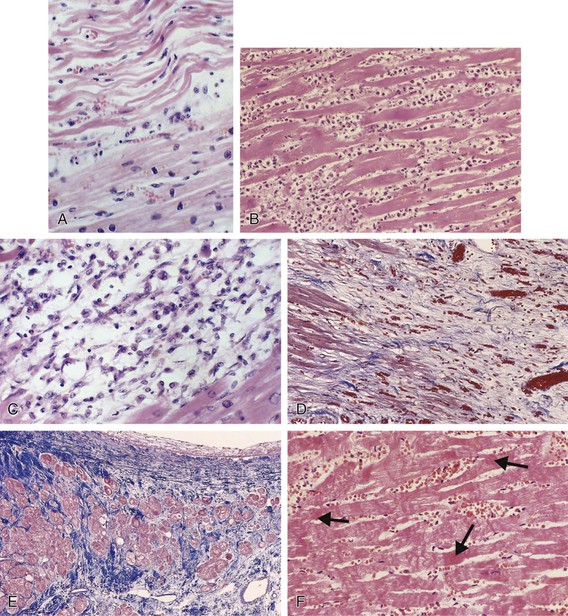
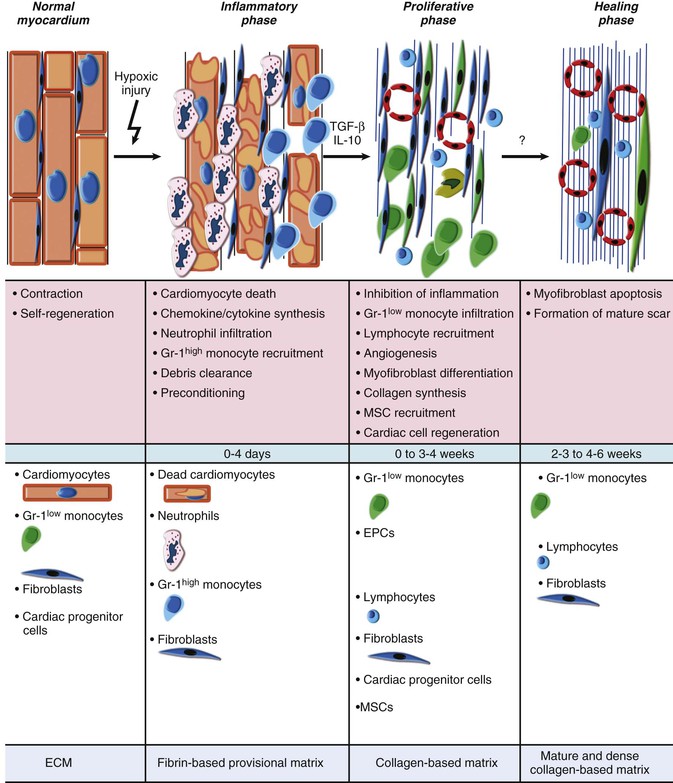
Current Concepts of the Cellular Events During Myocardial Infarction and Healing
Classic studies defined the sequence of cellular events that occur during human MI by careful histologic studies.37 Accumulation of granulocytes characterized the first days following MI. After the first days, mononuclear phagocytes accumulated in the infarct in tissue. Finally, granulation tissue characterized by neovascularization and accumulation of extracellular matrix (fibrosis) followed. Recent experimental work in mice has revealed a sequence of accumulation of subpopulations of mononuclear phagocytes.38 The first wave, occurring about days 1 to 3 following coronary ligation, consists of a proinflammatory subset of monocytes characterized by high proteolytic and phagocytic capacity and elaboration of proinflammatory cytokines. During a later phase (days 3 to 7), less inflammatory monocytes predominate and produce the angiogenic mediator vascular endothelial growth factor (VEGF) and the fibrogenic mediator transforming growth factor-beta (TGF-β). This highly orchestrated sequential recruitment of subpopulations of monocytes probably plays an important role in myocardial healing. The first wave of proinflammatory and phagocytically active mononuclear cells constitutes a “cleanup crew” that clears necrotic debris and paves the way for the second wave of less inflammatory monocytes, which contribute to healing by promoting the formation of granulation tissue.
Modification of Pathologic Changes by Reperfusion
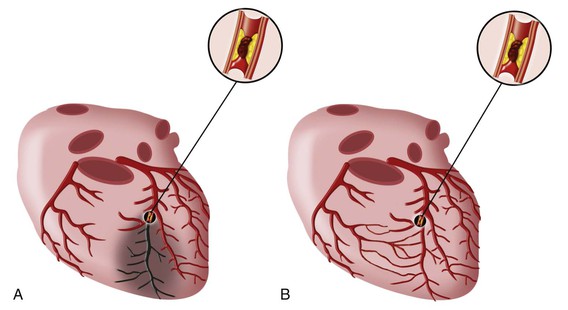
When reperfusion of myocardium undergoing the evolutionary changes from ischemia to infarction occurs sufficiently early (i.e., within 15 to 20 minutes), it can successfully prevent necrosis from developing. Beyond this early stage, the number of salvaged myocytes—and therefore the amount of salvaged myocardial tissue (area of necrosis/area at risk)—is directly related to the length of time of total coronary artery occlusion, the level of myocardial oxygen consumption, and collateral blood flow (Fig. 51-11). Reperfused infarcts typically show a mixture of necrosis, hemorrhage within zones of irreversibly injured myocytes, coagulative necrosis with contraction bands, and distorted architecture of cells in the reperfused zone (Fig. 51-12). Reperfusion of infarcted myocardium accelerates the washout of leaked intracellular proteins, thereby producing an exaggerated and early peak value of substances such as the MB fraction of creatine kinase (CK-MB) and cardiac-specific troponin T and I (see below).39
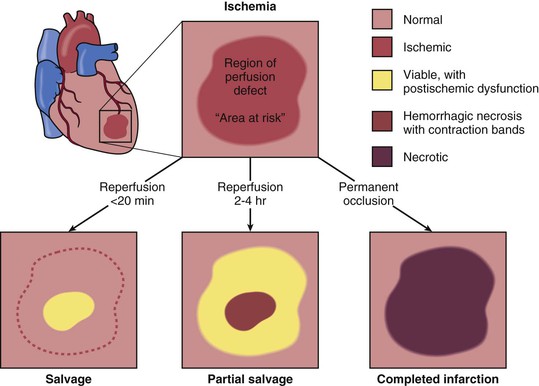
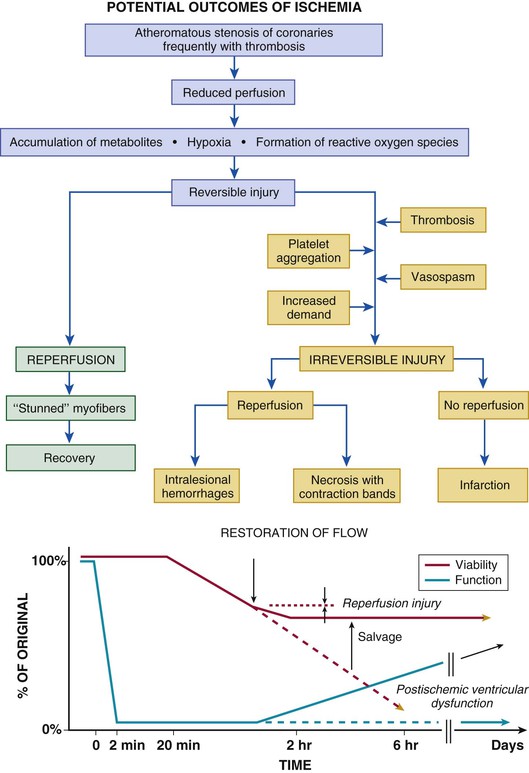
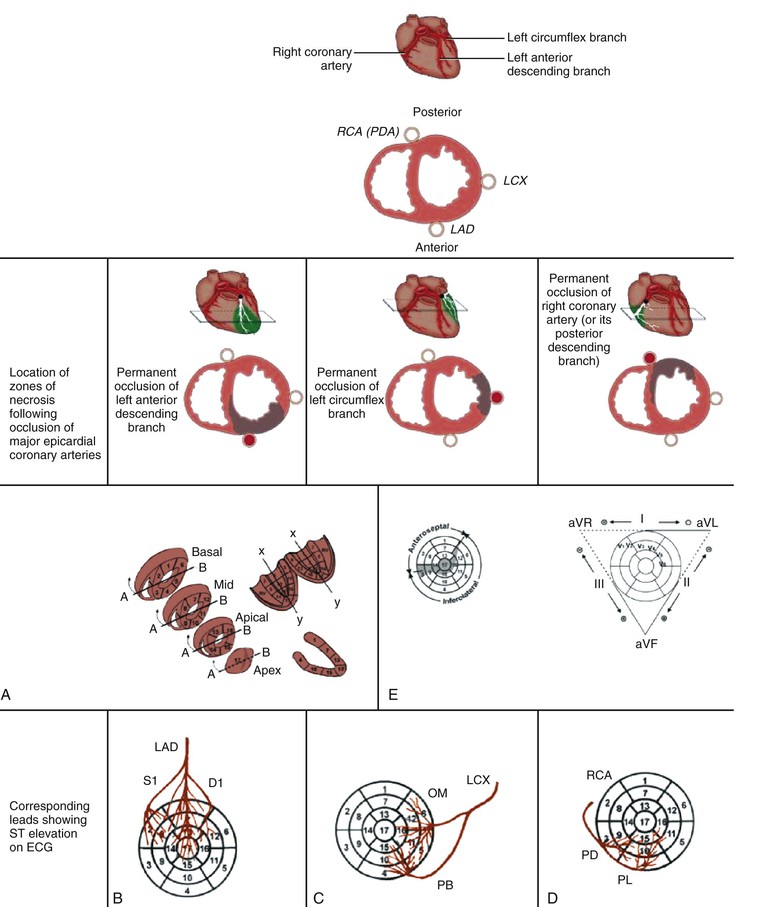
Coronary Anatomy and Location of Infarction
Angiographic studies performed in the earliest hours of STEMI have revealed an approximately 90% incidence of total occlusion of the infarct-related vessel. Recanalization as a result of spontaneous thrombolysis diminishes angiographic total occlusion in the period following the onset of MI. Pharmacologic fibrinolysis and PCI markedly increase the proportion of patients with a patent infarct-related artery early after STEMI.
A STEMI with transmural necrosis typically occurs distal to an acutely totally occluded coronary artery with thrombus superimposed on a ruptured plaque (see Fig. 51-5). Yet chronic total occlusion of a coronary artery does not always cause MI. Collateral blood flow and other factors such as the level of myocardial metabolism, the presence and location of stenoses in other coronary arteries, the rate of development of the obstruction, and the quantity of myocardium supplied by the obstructed vessel all influence the viability of myocardial cells distal to the occlusion. In many series of patients studied at necropsy or by coronary arteriography, a small number (5%) of those with STEMI have normal coronary vessels. An embolus that has lysed, a transiently occlusive platelet aggregate, or a prolonged episode of severe coronary spasm may cause the infarct in these patients.
Studies of patients in whom STEMI ultimately develops after having undergone coronary angiography at some time before its occurrence have helped clarify the coronary anatomy before infarction. Although high-grade stenoses, when present, more frequently lead to STEMI than do less severe lesions, most occlusions occur in vessels with a previously identified stenosis of less than 50% on angiograms performed months to years earlier.27 This finding supports the concept that STEMI results from sudden thrombotic occlusion at the site of rupture of previously nonobstructive but lipid-rich plaque. When collateral vessels perfuse an area of the ventricle, an infarct may occur at a distance from a coronary occlusion. For example, following gradual obliteration of the lumen of the right coronary artery, collateral vessels arising from the left anterior descending coronary artery can keep the inferior wall of the left ventricle viable. Later, an occlusion of the left anterior descending artery may cause infarction of the diaphragmatic wall.
Right Ventricular Infarction
Approximately 50% of patients with inferior infarction have some involvement of the right ventricle.40 Among these patients, right ventricular (RV) infarction occurs exclusively in those with transmural infarction of the inferoposterior wall and the posterior portion of the septum. RV infarction almost invariably develops in association with infarction of the adjacent septum and inferior left ventricular (LV) walls, but isolated infarction of the right ventricle is seen in just 3% to 5% of autopsy-proven cases of MI. RV infarction occurs less commonly than would be anticipated from the frequency of atherosclerotic lesions involving the right coronary artery. The right ventricle can sustain long periods of ischemia but still demonstrate excellent recovery of contractile function after reperfusion.
Atrial Infarction
This type of infarction occurs in up to 10% of patients with STEMI if PR-segment displacement is used as the criterion. Although isolated atrial infarction is observed in only 3.5% of patients with STEMI at autopsy, it often occurs in conjunction with ventricular infarction and can cause rupture of the atrial wall.41 This type of infarction is more common on the right side than on the left side, occurs more frequently in the atrial appendages than in the lateral or posterior walls of the atrium, and can result in thrombus formation. Atrial infarction is frequently accompanied by atrial arrhythmias and has been linked to reduced secretion of atrial natriuretic peptide and to a low–cardiac output syndrome when RV infarction coexists.
Collateral Circulation in Acute Myocardial Infarction (See Chapter 49)
The coronary collateral circulation is particularly well developed in patients with coronary occlusive disease, especially with reduction of the luminal cross-sectional area by more than 75% in one or more major vessels; in patients with chronic hypoxia, as occurs in cases of severe anemia, chronic obstructive pulmonary disease, and cyanotic congenital heart disease; and in patients with LV hypertrophy (see Fig. 51-6).42
The magnitude of coronary collateral flow is a principal determinant of infarct size. Indeed, patients with abundant collateral vessels commonly have totally occluded coronary arteries without evidence of infarction in the distribution of that artery; thus survival of myocardium distal to such occlusions depends largely on collateral blood flow. Even if the collateral perfusion existing at the time of coronary occlusion does not prevent infarction, it may still exert a beneficial effect by preventing the formation of LV aneurysms. The presence of a high-grade stenosis (90%), possibly with periods of intermittent total occlusion, probably permits the development of collateral vessels that remain only as potential conduits until a total occlusion occurs or recurs. Total occlusion then brings these channels into full operation. Patients with angiographic evidence of collateral formation have improved angiographic and clinical outcomes after MI.
Nonatherosclerotic Causes of Acute Myocardial Infarction
Numerous pathologic processes other than atherosclerosis can involve the coronary arteries and result in STEMI (see Table 51-3). For example, coronary arterial occlusions can result from embolization of a coronary artery. The causes of coronary embolism are numerous: infective endocarditis and nonbacterial thrombotic endocarditis (see Chapter 64), mural thrombi, prosthetic valves, neoplasms, air introduced at the time of cardiac surgery, and calcium deposits from manipulation of calcified valves at surgery. In situ thrombosis of coronary arteries can occur secondary to chest wall trauma (see Chapter 72).
A variety of inflammatory processes can cause coronary artery abnormalities, some of which mimic atherosclerotic disease and may predispose to true atherosclerosis. Epidemiologic evidence suggests that viral infections, particularly with Coxsackie B virus, may uncommonly cause MI. Viral illnesses occasionally precede MI in young persons who are later shown to have normal coronary arteries.
Syphilitic aortitis can produce marked narrowing or occlusion of one or both coronary ostia, whereas Takayasu arteritis can result in obstruction of the coronary arteries. Necrotizing arteritis, polyarteritis nodosa, mucocutaneous lymph node syndrome (Kawasaki disease), systemic lupus erythematosus (see Chapter 84), and giant cell arteritis can cause coronary occlusion. Therapeutic levels of mediastinal radiation can result in coronary arteriosclerosis43 with subsequent infarction. MI can also result from coronary arterial involvement in patients with amyloidosis (see Chapter 65
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


