Figure 11–1.
Schematic representation of the voltage gated Na+ channel α(alpha)-subunit and β(Beta)-subunits. (a) The α(alpha)-subunit consists of four domains (D1–D4), each composed of six membrane-spanning segments (S1–S6) linked by intracellular and extracellular loops. The linkers between S5 and S6 control ion selectivity and permeation of the channel, while the positively charged segments S4 (in pink) act as a voltage sensor. Differently coloured circles display the location of mutations associated with Brugada syndrome, Long QT syndrome type 3, sudden infant death syndrome, cardiac conduction disease, sick sinus syndrome/atrial standstill, dilated cardiomyopathy, atrial fibrillation and overlap syndrome [1–12]. DI domain I, DII domain II, DIII domain III, DIV domain IV. (b) The β(Beta)-subunits consist of an extracellular N terminus, one transmembrane segment, and an intracellular C terminus. Note that the β(Beta)1-subunit has two isoforms. A mutation found in cardiac conduction disease was located in the β(Beta)1-subunit, whereas a mutation in Brugada syndrome was located in the β(Beta)1B subunit [10, 13–16] (The authors thank Dr. Andre Linnenbank for providing Fig. 11.1)
The SCN5A gene encodes Nav1.5, the α(alpha)-subunit of the cardiac sodium channel. Nav1.5 consists of an intracellular N-terminus, four homologous domains (DI–DIV), intracellular linkers between these domains, and the C-terminus. Each domain consists of six transmembrane segments (S1–S6). Segment 5 (S5) and S6 are positioned close to the pore, so that they form a pore lumen where sodium ions can traverse the sarcolemma. S4 is positively charged and plays a role as the channel’s voltage sensor. The P-loop between S5 and S6 lines the pore of the channel extracellularly.
At the normal resting membrane potential of working myocytes and myocytes of the specific conduction system (−90 mV), the sodium channel remains closed. When excitatory current from adjacent cells arrives and depolarizes the membrane potential positive to −75 mV, the sodium channel’s activation threshold, all four S4 segments (the voltage sensors) move outward and the channels are activated, which leads to the opening of the channels. At the same time, the fast inactivation starts using the DIII-DIV linker as a ‘lid’ that occludes the pore intracellularly. The C-terminus helps to stabilize the fast inactivation process [18]. Since the inactivation process is slower than the activation process, channels remain open transiently during phase 0 of the action potential. Slow inactivation follows and, by the end of phase 1, virtually all sodium channels are inactivated. The remaining sodium current, the so-called ‘late sodium current (Isus)’ is still recorded during the repolarization phase of the action potential and slowly inactivates. The conformational change of the sodium channel during slow inactivation is less known, but there are reports suggesting the involvement of the voltage sensors (S4), S5–S6 linkers, the S6 segments or the C-terminus [19–24]. After a few hundred milliseconds to a few seconds, the sodium channels are completely inactivated and wait for the next stimulation for activation.
Nav1.5 itself can generate sodium currents when heterologously expressed alone, but β(Beta)-subunits play important roles in modulating current density and gating of the sodium channels. There are four β(Beta)-subunits (β(Beta)1-β(Beta)4) in the heart and they are encoded by the SCN1B-4B genes [25–27]. Beta-subunits consist of an extracellular N-terminus, a single transmembrane segment, and an intracellular C-terminus. Studies using both α(alpha)- and β(Beta)-subunits have shown that β(Beta)-subunits interact with the α-subunit and modify the expression of α(alpha)-subunit in the cell membrane or its gating process [28–31]. Besides β(Beta)-subunits, there are regulatory proteins, such as glycerol-3-phosphate dehydrogenase like protein (GPD1L), multicopy suppressor of gsp1 (MOG1), and caveolin-3 (CAV3), which are shown to interact with Nav1.5 [17].
Mutations in the α(alpha)-subunits, β(Beta)-subunits and some regulatory proteins of the cardiac sodium channel produce dysfunctional sodium channels and cause diseases, such as BrS, LQTS, PCCD, atrial standstill, SSS, and DCM through either gain-of-function or loss-of-function of the sodium channel (Table 11.1) [32].
Table 11–1.
Current changes in sodium ion channelopathies
Primary electric disease | Reported changes in INa |
|---|---|
Brugada syndrome | ↓ |
Long QT syndrome | ↑ |
Progressive cardiac conduction disease | ↓ |
Sick sinus syndrome | ↓ |
Atrial fibrillation | ↓, ↑ |
Sudden infant death syndrome | ↓, ↑ |
Dilated cardiomyopathy | ↓ |
Interestingly, most mutations reported in β(Beta)-subunits are located in the N-terminus, which suggests that this region is involved in the regulatory role of the β(Beta)-subunits in the sodium currents. Importantly, whether a mutation found in a patient is disease-causing or not needs careful interpretation, because putative causative mutations in the α(alpha)- and β(Beta)-subunits of the cardiac sodium channel are found mostly in a single index case without sound linkage data. Besides, rare SCN5A variants may be found in the general population at a proportion as high as 2–8 % [1, 2].
Sodium Ion Channelopathies
Brugada Syndrome (BrS)
BrS is an inherited disease which is characterized by coved type ST elevation in the right precordial leads of the ECG (Fig. 11.2), and sudden cardiac death (SCD) due to ventricular fibrillation (VF). VF predominantly occurs at rest or during sleep. Fatal arrhythmias have an age of onset in the 40s on average and predominantly occur in males [33]. In patients with BrS, 20–50 % have a family history of SCD. BrS has an autosomal dominant pattern of transmission with incomplete penetrance and variable expressivity [33]. SCN5A mutations are found in 20 % of BrS patients and, although rare, mutations in other genes have also been reported. These genes encode various calcium channels (CACNA1C, CACNB2b, CACNA2D1), β(Beta)-subunits of sodium channels (SCN1B, SCN3B), proteins which affect trafficking of sodium channels (GPD1L, MOG1), and the transient outward potassium current, Ito (KCNE3, KCND3, KCNE5) [34]. Risk stratification is difficult and, although BrS is an inherited disease, a SCN5A mutation or family history of SCD has not been consistently found to be a risk marker for SCD [35–37].
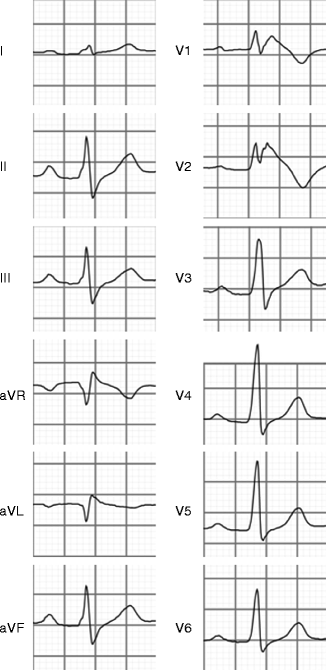
Figure 11–2.
Diagnostic ECG of Brugada syndrome (type 1 ECG). Note that ST segment elevation with high take-off (J point) of ≥2 mm and a subsequent negative T wave is observed in the right precordial leads V1–V2
More than 200 SCN5A mutations have been reported in BrS as a putative causal mutation so far [1]. A recent multicenter study by Kapplinger et al. analyzed 2,111 unrelated patients with variable ethnic backgrounds who were referred for genetic analysis of SCN5A. This study found 293 possibly causal mutations, of which 77 % were found only once. The most frequently found mutations in multiple cases were E1784K, F861WfsX90, D356N and G1408R. Two-thirds of mutations reported in the study were missense mutations, and the rest were frameshift mutations, nonsense mutations, splice-site mutations and in-frame insertions or deletions. Seventy-one percent of the mutations were located in one of the four transmembrane domains [1].
Experimental studies in heterologous expression systems have shown that BrS-related mutations cause loss-of-function of the sodium channel through decreased expression of sodium channels in the sarcolemma [38], expression of non-functional channels [3] or altered gating properties (delayed activation, earlier inactivation, faster inactivation, enhanced slow inactivation and/or delayed recovery from inactivation) (Table 11.2) [4, 39–42]. Of note, less than 10 % of the reported mutations found in BrS underwent functional analysis and the relationship between a mutation and clinical symptoms is often not well-understood. So far, there is only one study which suggested that truncated Nav1.5 proteins (due to nonsense mutations, frameshift mutations or in-frame insertions or deletions) or missense mutations with >90 % peak INa reduction may lead to severe conduction disturbance or syncope [43], but it is presently too early to conclude if the findings are relevant or not.
Table 11.2
Reported biophysical mechanisms of increase in net sodium current (gain-of-function) and reduction in net sodium current (loss-of-function)
Gain of function |
Persistent current (disruption of fast inactivation) |
Changes in voltage dependence of activation (hyperpolarizing shift) and inactivation (depolarizing shift) |
Faster recovery from inactivation |
Slower inactivation |
Loss of function |
Reduction in current density |
Reduced number of functional sodium channels in sorcolemma |
Mutation located in ion-conducting pore |
Truncated protein due to premature stop codon |
Trafficking defect (channel protein retention in endoplasmic reticulum) |
Gating changes |
Changes in voltage dependence of activation (depolarizing shift) and inactivation (hyperpolarizing shift) |
Slower recovery from inactivation |
Enhanced intermediate inactivation |
Enhanced closed-state inactivation |
Interestingly, not only putative causative SCN5A mutations have been reported to modify clinical phenotypes of BrS, but also SCN5A polymorphisms. H558R, a common SCN5A polymorphism, was shown to modify the electrophysiological property of a BrS-related mutant sodium channel as well as BrS type 1 ECG [44]. Possibly explaining, at least in part, the role of different ethnic background in the clinical effect of SCN5A, an SCN5A promoter polymorphism in a haplotype variant with a relatively high prevalence was reported to lead to difference in cardiac conduction parameters in individuals of Asian origin [45]. A study in two different strains of transgenic mice (129P2 and FVB/N) carrying the same SCN5A mutation (1798insD/+) showed that ventricles in 129P2 mice expressed low to undetectable sodium channel auxiliary subunit β(Beta)4, which led to slower conduction in the right ventricle in 129P2 mice compared to FVB/N mice [46].
All these studies suggest that phenotypes related to SCN5A mutations and/or BrS may be affected by multiple factors. Importantly, one study on large SCN5A positive families with clinically affected BrS family members showed that, in the same family, there were individuals with BrS phenotype (type1 ECG) without the familial SCN5A mutation [47]. Therefore, a single SCN5A mutation may not play a major role to cause BrS.
Mutations in genes such as auxiliary β(Beta)-subunits of sodium channels (SCN1B, SCN3B) or GPD1L and MOG1, which affect trafficking of sodium channels, have been reported in relation to BrS [13, 14, 48–54]. Cellular electrophysiological studies have shown that mutations in SCN1B, SCN3B, GPD1L or MOG1 led to loss-of-function of INa. However, GPD1L is the only gene with sound genetic linkage so far and all others have been identified with candidate gene analysis used in single patients or in small families. Therefore, further study is needed to elucidate the pathophysiologic mechanism of BrS and the role of cardiac sodium channel-related genes or proteins [55].
Recent studies showed that structural abnormalities exist in some patients with BrS [56, 57]. As the derangements of both BrS and arrhythmogenic right ventricular cardiomyopathy (ARVC) are anatomically located predominantly in the right ventricle, overlap between BrS and ARVC has been suggested [58–61]. From a genetic point of view, there are few reports [62, 63]. One study has reported a SCN5A mutation in a single patient of ARVC with a drug-induced type-1 BrS-ECG [63]. Therefore, it is not clear at this point if these two conditions may share the same mechanism or not.
Long QT Syndrome (LQTS)
LQTS is characterized by QT prolongation on the baseline ECG and has a high risk of SCD due to torsade de pointes ventricular tachycardia, which often degenerates into VF. LQTS is classified into two forms: congenital or acquired. Acquired LQTS is precipitated by drug use and/or electrolyte imbalance such as hypokalemia, hypocalcemia or hypomagnesemia. Congenital LQTS is a hereditary disease and estimated to affect 1/5,000 individuals with either an autosomal-dominant (Romano-Ward syndrome) or autosomal-recessive inheritance pattern (Jervell and Lang-Nielsen syndrome) [64]. Thus far, 13 different types of LQTS, each based on a different causal gene, have been reported. Most types are related to cardiac potassium channels and a few are related to genes which encode the calcium channel or the sodium channel [65]. LQTS related to a mutation in the SCN5A gene is named LQTS type 3 (LQT3). The prevalence of LQT3 among genotyped LQTS patients is 13 %, whereas the prevalence of LQT1 (KCNQ1 mutation) and LQT2 (KCNH2 mutation) are 43 and 32 %, respectively [2]. The other subtypes are rare.
Patients with LQT3 experience significantly more severe arrhythmic events compared to LQT1 or LQT2 [66]. Events are related to bradycardia and occur at rest or during sleep [67]. Beta blockers appear to be effective in preventing cardiac events, but seem less effective than in LQT1 or LQT2 [67–69]. Other therapeutic approaches in LQT3 include antiarrhythmic drug therapy using mexiletine, flecainide or ranolazine [70–73], although the therapeutic efficacy of mexiletine may be mutation-specific and can lead to QT prolongation and arrhythmic events in other cases [74].
So far, more than 70 SCN5A mutations have been reported in LQT3 and most of them are missense mutations. Of note, whether a missense mutation is pathogenic depends on its location within the sodium channel [75]. Functional analysis in heterologous expression systems was performed in approximately half of the published mutations [76]. These studies have consistently shown that these mutations lead to gain-of-function of the sodium channel. This is due to the increase of Isus, caused by disruption of the fast inactivation process [77], increasing window current [78, 79], slower inactivation [78, 80], faster recovery from inactivation [81, 82] or larger peak INa density (Table 11.2) [83]. In rare cases, mutations in genes such as CAV3, SCN4B, or α(alpha)1 syntrophin (SNTA1) have been also shown to cause LQTS through increased Isus [15, 84–86].
Progressive Cardiac Conduction Disease (PCCD)
PCCD, also called Lenègre or Lev’s disease, is originally characterized by progressive impairment of the conduction system with widening of the QRS complexes. No structural abnormality is observed and complete atrioventricular (AV) block may occur as a consequence, which necessitates pacemaker implantation. Histopathologic studies revealed a sclerodegenerative process in the His-Purkinje system [87, 88], and the disease was considered a primary degenerative disease or an exaggerated aging process. Patients are usually asymptomatic, but may experience syncope or SCD when advanced or complete AV block occurs.
From a genetic perspective, Schott et al. were the first to report a large family with cardiac conduction disease related to a SCN5A mutation [89]. Functional studies in families with cardiac conduction disease revealed loss-of-function of the sodium channel [90, 91]. As further research on SCN5A mutations was performed, it has been revealed that a single SCN5A mutation may show overlap of several phenotypes such as BrS, LQTS, atrial fibrillation, SSS, and DCM in combination with conduction disturbances [4–6, 92], (details discussed in the paragraph Overlap syndrome). Of note, the severity of clinical phenotypes varies and it is not known which factors other than SCN5A mutations cause more severe conduction disease with earlier disease onset or lead to different phenotypes. As the penetrance and phenotypic expression of a SCN5A mutation vary, further research to elucidate the mechanism of PCCD related to SCN5A mutations including (genetic) modifying factors is necessary.
Sick Sinus Syndrome (SSS)
SSS is a disease caused by sinoatrial node dysfunction and/or conduction disturbances between the sinoatrial node and surrounding atrial tissues. Clinically, sinus bradycardia, sinus arrest or Brady-Tachy syndrome is observed. It is usually diagnosed in the elderly and patients show symptoms such as shortness of breath, fatigue, palpitations or syncope. Patients with SSS are treated with pacemaker implantation. In hereditary SSS, loss-of-function SCN5A mutations have been reported. A study on congenital SSS cases has shown compound heterozygous mutations in SCN5A [93]. Loss-of-function mutations in SCN5A found in SSS may lead to overlap with other phenotypes, where some family members exhibit SSS, while others present with BrS or PCCD [6, 7, 92]. The mechanism of SSS due to loss-of-function mutations may be sinoatrial exit block by disruption of conduction into the surrounding atrium. On the other hand, bradycardia and sinus node dysfunction coexisting in LQT3 families is caused by gain-of-function of the sodium channel [94]. In this study, it was experimentally explained that the presence of Isus (1–2 % of peak INa) and negative shift in voltage dependence of inactivation may reduce sinus rate by up to 10 %.
Atrial standstill has also been related to a SCN5A mutation. In this study, a loss-of-function SCN5A mutation in combination with a loss-of-function connexin-40 polymorphism were described as a cause of atrial standstill (Fig. 11.3) [95].
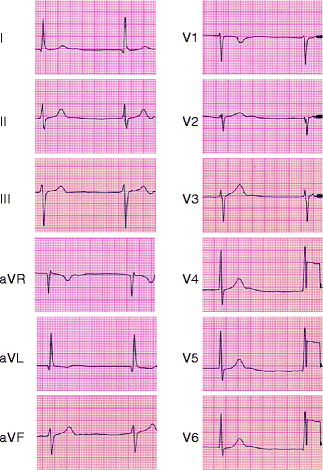
Figure 11–3.
ECG of atrial standstill in a patient with SCN5A D1275N and a Cx40 polymorphism. Junctional rhythm is observed
Atrial Fibrillation (AF)
AF is recognized as the most common cardiac dysrhythmia and a major cause of morbidity and mortality. Predisposing conditions include aging, diabetes, obesity, heart failure, coronary and valvular disease and prior cardiac surgery [99]. In a minority of cases, AF occurs without the aforementioned conditions. As a rare subtype, a familial form of AF has been recognized for many years. Heritable AF has been identified using linkage analysis [100, 101]. Mutations in potassium channels, sodium channels (α(alpha)- and β(Beta)-subunits), nucleoporin, connexin-40 and atrial natriuretic peptide have been reported in relation with AF [102]. Recent extensive research on ion channelopathies also revealed that AF was observed in BrS or LQTS [103–105].
Regarding the sodium channelopathies, mutations in the genes encoding either the α(alpha)-subunit or the β(Beta)-subunits of the cardiac sodium channel have been reported in AF, but appear to be rare [8, 16, 106, 107]. The first SCN5A mutations related to AF were found in DCM patients [9]. Subsequently, another SCN5A mutation was reported in a family with lone AF [8]. A common polymorphism (H558R), known as a modifier to reduce INa in heterologous expression systems, was also found more frequently in lone AF patients compared to controls [106]. These mutations are considered to create conduction delay and predispose to reentry in the atrium through loss-of-function of INa. Interestingly, gain-of-function type mutations were also reported in AF [5, 108]. These mutations have shown to delay inactivation, and therefore could lead to AF by increasing atrial excitability.
Sudden Infant Death Syndrome (SIDS)
SIDS is defined as the sudden death of an infant younger than 1 year of age, which remains unexplained after a thorough investigation, including a complete autopsy, clinical history, and death scene investigation. Risk factors identified by epidemiological studies include prone or side positions for infant sleep, soft bedding and sleep surfaces, and overheating [109].
Genetic factors have been also found to play a role in some patients with SIDS. Since 1970s, the association between congenital LQTS and SIDS has been pointed out [110, 111] and it is currently estimated that 5–20 % of SIDS are related to ion channelopathies [10, 112]. So far, 13 cardiac channelopathy susceptibility genes have been implicated in the pathogenesis of SIDS. These include potassium channel genes (KCNQ1, KCNH2, KCNE1, KCNE2, KCNJ8), sodium channel genes and related regulatory proteins (SCN5A, SCN3B, SCN4B, GPDL1, CAV3, SNTA1), the calcium release channel of the sarcoplasmic reticulum (RYR2), and other genes (GJA1) [31, 113–125]. The clinical syndromes associated with these mutations are LQTS, short QT syndrome, BrS, catecholaminergic polymorphic ventricular tachycardia or idiopathic ventricular fibrillation. Of these clinical phenotypes, LQTS is the most extensively studied in the newborn. In a prospective study of a large cohort of 34,000 neonates with an ECG performed on the third or fourth day of life, the authors demonstrated that 50 % of SIDS infants showed a prolonged QT interval [126]. The same group subsequently performed genetic testing which resulted in finding genes related to LQTS [127, 128]. More recently, they reported that the prevalence of LQTS disease-causing mutations in SIDS was 9.5 % [129].
Currently, SIDS related to mutations in cardiac sodium channel genes (SCN5A, SCN3B, SCN4B) constitutes 43 % of all mutations found and the majority are missense mutations [10]. Functional analysis of several mutations has revealed that some mutations cause gain-of-function, while others cause loss-of-function. Clinically, this leads to the LQT3 and BrS phenotypes, respectively. Another mutation found in SCN3B (V36M) showed both gain-of-function and loss-of-function phenotypes secondary to decreased peak INa and increased late INa. The SCN5A polymorphism S1103Y, common in African Americans, has also been implicated as a possible modifier to increase susceptibility to SIDS by increasing late INa [130].
Although the mechanism of SIDS is not fully elucidated, some potential SIDS victims will certainly benefit from genetic analysis, considering that the prevalence of channelopathies in SIDS is up to 20 %. Genetic testing in SIDS victims with a channelopathy may also help to evaluate the variable expressivity in the ion channelopathies.
Dilated Cardiomyopathy (DCM)
DCM is diagnosed when there is evidence of dilatation and impaired contraction of the left ventricle or both ventricles. The disease is considered idiopathic if other causes (coronary artery disease, active myocarditis, hypertension, cardiotoxic drugs such as doxorubicin, alcohol abuse) are excluded. Familial DCM accounts for >20 % of idiopathic DCM cases [131], but mutations are found only in 30–35 % [132]. Most of the genes reported in DCM encode sarcomeric proteins [132]. A recent study using a large multicenter DCM cohort has shown that 1.7 % of 338 DCM subjects (including 289 probands) carried a mutation in SCN5A [133]. Of 15 SCN5A mutation carriers in this study, 14 (93 %) manifested arrhythmias (supraventricular arrhythmia, SSS, ventricular tachycardia, and PCCD). Two-thirds of the reported SCN5A mutations (six of nine) were located in S3 and S4 transmembrane segments. This provides a clue to a shared mechanism of SCN5A-related DCM. The most frequently reported mutation in DCM so far is D1275N. It has been disputed whether DCM with SCN5A D1275N occurs as a consequence of atrial arrhythmias, because atrial arrhythmias frequently appear as the first clinical phenotype in the 20s to 30s, while DCM develops later in life [134]. To answer this question, Watanabe et al. studied SCN5A-D1275N using a mouse model and a heterologous expression system [135]. They showed that dysfunction of the conduction system, atrial and ventricular tachyarrhythmias, and DCM phenotype was observed in mice that carried the SCN5A-D1275N mutation. Yet, studies in the heterologous expression system did not reveal major differences between wild-type and D1275N. Accordingly, the true nature of SCN5A mutations may only be unmasked if different expression systems are used in experimental studies. For other SCN5A mutations found in DCM [9, 133], further studies are awaited to prove if they are causal for DCM or not.
Overlap Syndrome
In overlap syndrome, a single SCN5A mutation is associated with multiple phenotypes in a single patient, in a family sharing the same mutation or in different families having the same mutation. For example, loss-of-function mutations in SCN5A have been shown to present multiple phenotypes, such as BrS, SSS or PCCD [3, 6, 92, 94]. More remarkably, some SCN5A mutations are associated with multiple phenotypes which are associated both with loss-of-function of the sodium channel (BrS) and gain-of-function (LQTS) [4, 5, 7]. This appears to be contradictory from a functional point of view. Patch-clamp studies of SCN5A 1795insD showed that this may be explained by concurrent gating changes: enhanced late sodium current leads to gain-of-function and LQT3, whereas faster intermediate inactivation causes loss-of-function and, as a consequence, BrS. A subsequent study showed that a mutation of the same residue to a histidine (Y1795H) or cysteine (Y1795C) also showed distinct phenotypes: BrS and LQT3, respectively [136].
Why other single SCN5A mutations may lead to different clinical phenotypes is not fully understood. However, some studies suggested that the existence of modifier genes affects the phenotype. For example, the D1275N mutation leads to atrial standstill when combined with a connexin-40 polymorphism, but to DCM and conduction disturbances when such a polymorphism is absent [95, 137, 138]. A common SCN5A polymorphism, H558R, was also reported to modify the expression of a SCN5A mutation in an experimental study, or the clinical severity of conduction disease or BrS [44, 139, 140]. Different clinical expressivity of a SCN5A mutation in different ethnic groups may be also explained by different genetic backgrounds. Bezzina et al. reported that a promoter haplotype variant of the sodium channel was commonly found in Asian populations, but not in Caucasians. This variant led to conduction disturbance represented by ECG parameters, such as longer PR and QRS duration. Gender also plays a role in different phenotypic expressivity. For example, in BrS, it is known that men are predominantly clinically affected. One of the reasons is that action potential properties are different between men and women. Ito is more abundant in men, whereas ICaL is more expressed in epicardium of women [141–143]. Testosterone is another modifier that has been reported to affect BrS type 1 ECG [144]. Variants in KCNE5, which is located in the X chromosome, have been recently reported to be implicated as a modifier in BrS or idiopathic ventricular fibrillation [54].
Overlap syndrome suggests that there are several modifiers including gene mutations/variants affecting the expressivity of phenotypes related to SCN5A mutations. Further study is awaited to reveal the mechanisms leading to different clinical phenotypes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


