Fig. 8.1
Conceptual model linking social stress to lung growth/development and asthma
Early Life Programming of Lung Structure and Function
Exposure to negative social factors (e.g., low SES, psychological stress) during prenatal and/or early childhood development may alter the normal course of lung morphogenesis, resulting in changes that affect both structure and function of the respiratory system [17, 18]; this is referred to as developmental plasticity. The fetal and infant respiratory system may be particularly vulnerable, in part, due to the long developmental and maturation period of the lung [19]. Lung development starts in utero and progresses through a series of carefully orchestrated stages starting with establishment of core lung structure with airway branching in the embryonic (4–7 weeks gestation) and pseudoglandular (5–17 weeks) stages of early lung development. This is followed by the canalicular stage (16–24 weeks) during which air spaces are beginning to open with subsequent formation of saccular units in the airways during the saccular stage (24–35 weeks); secondary septa divide the saccular units during the alveolar stage which starts late in pregnancy (beginning at ~34 to 36 weeks gestation) and continues postnatally over a period of several years. Over the first 2–3 years of life, the full complement of 23 airway generations and approximately 300 million alveoli are formed. Throughout early lung development, rapidly proliferating cells are most susceptible to the adverse effects of social and physical environmental factors that may impact many physiological systems underlying respiratory developmental processes [20]. Finally, there is a prolonged period of equilibrated lung growth that continues until body growth stops in late adolescence and early adulthood [21]. Gross changes in lung structure occur if development is disrupted during the embryonic phase which is characterized by a period of rapid cellular differentiation and morphogenesis. When late lung development is disrupted, lung architecture is malformed with consequent lung function changes. The underlying mechanisms leading to reduced lung function and exaggerated airway responsiveness involve chronic airway inflammation associated with a cycle of injury, repair, and remodeling [22, 23]. Furthermore, the fundamental cause of the airway inflammation is aberrant and/or excessive immune responses to various social and physical environmental factors [22] and the most common cause of chronic airway inflammation in early childhood is arguably asthma. Notably, airway inflammation and remodeling begin and progress even in the presymptomatic state in early childhood [24, 25]. Given this and the fact that most of the extant research on social disparities in respiratory disease has centered on asthma [26–29], many of the principles presented herein will highlight asthma albeit they are more broadly applicable to other respiratory disorders.
Programming Stress Pathways Involved inRespiratory Disease
Critical Periods of Development and Perinatal Programming
It is essential to characterize mechanisms that lead to and maintain early predisposition if we want to identify individuals at risk for chronic respiratory disorders. Most respiratory conditions, including asthma and other chronic conditions, have their origin in early life. Immune and lung development occur largely in utero and during early childhood. Research continues to delineate early immunophenotypes and early airway response outcomes among children predisposed to asthma (atopic and nonatopic) and other chronic atopic disorders [30, 31]. Regulatory pathways that involve the collaboration of innate and adaptive immune responses are involved. Influences of factors outside the immune system, i.e., neurohormonal, autonomic nervous system (ANS), and mitochondrial function may also be involved. Plasticity is a consequence of environmental exposures (both social and physical) in critical periods affecting key physiological systems involved in developmental processes. Although both asthma and respiratory function are polygenic traits [32, 33], maternal factors in particular contribute to the intergenerational correlation [32, 34, 35]. That is, the risk of developing asthma is particularly increased if a positive parental history of atopy is present with effects being strongest for maternal history [34, 35]. Studies have also shown a greater correlation in forced expiratory volume in 1 s (FEV1) and other lung function parameters between mothers (compared with fathers) and offspring [32]. In addition to heritable traits , this may be due to perinatal programming—the influence of nongenetic or environmental factors in the perinatal period that organizes or imprints physiological systems. There is a growing list of environmental factors that may have programming effects including stress-induced changes in a number of molecular, cellular, and physiological states and their interrelating systems over development. As summarized below, in response to chronic stress, physiological systems may function at higher or lower levels than during typical homeostasis resulting in systems being permanently organized toward trajectories of poorer lung function and enhanced respiratory disease risk.
Immune System
Central to the pathophysiology of asthma phenotypes, as well as other respiratory diseases, are mechanisms of inflammation. These mechanisms overlap and may include immune-mediated inflammation associated with a Th2-biased response [36] and a tendency to produce immunoglobulin E (IgE) in response to environmental stimuli (e.g., allergens). The Th1–Th2 paradigm involves a complex interaction of T and B lymphocytes, resulting in the production of higher levels of particular cytokines, such as interleukin-4 (IL-4) or IL-13 and the more recently described IL-9, IL-25, and IL-31 as well as lower levels of interferon-γ (IFN-γ) [37]. Evidence suggests that those with early (i.e., starting in the first 2–3 years) sensitization to allergens are at greatest risk of developing chronic atopic disorders, airway inflammation, and obstruction [as reviewed in [38]]. While this paradigm has been useful in understanding the large fraction of subjects with allergic asthma, it is now recognized that the Th2-biased polarization of adaptive immunity is likely only one of numerous axes that give rise to heightened susceptibility to airway inflammation and altered reactivity [39]. Antigen-independent responses involving innate immune cells (e.g., bronchial epithelial cells, alveolar macrophages, and dendritic cells) may also be important [40, 41] and include novel cytokines (e.g., IL-17) [42]. Factors, including stress [41, 43], that disrupt maturation of local immune networks (e.g., dendritic cells [DCs], epithelial cells [ECs], regulatory T cells) may predispose to ongoing eosinophilic and neutrophilic inflammation.
These immune mechanisms have their roots in utero with an immunological bias toward a Th2 phenotype [44–50]. Immune programming can also be influenced by early postnatal environmental factors [51, 52]. Consequently, researchers have begun to examine in vitro responses of peripheral blood mononuclear cells (pBMCs) to allergens or mitogens to gain a better understanding of the immunodeviations that facilitate the manifestation of asthma and atopy in response to environmental factors [30]. The influence of stress on the timing and trajectory of these immunophenotypes and their relationship to the later development of clinical disorders, however, has only just begun to be studied. Prospective epidemiological studies have linked early life caregiver stress to dysregulation of immune function in a birth cohort predisposed to allergy, i.e., greater antigen-specific TNF-alpha production [53] and cord blood total IgE [54–56]. Interestingly, it has also been demonstrated that increased prenatal maternal stress is associated with increased IL-8 and TNF-α production following microbial stimulation suggesting that stress may operate through Toll-like receptor-dependent pathways [57]. Furthermore, low maternal childhood SES appears to be important for immune regulation and risk of adverse respiratory outcomes in children suggesting possible intergenerational effects [7].
Neuroendocrine and Autonomic Nervous Systems
Both glucocorticoid (GC) action and sympathovagal balance play a role in immunomodulation as well as fetal and postnatal lung development [38, 43, 58]. While research has made considerable strides toward the advancement of immune function assessment in the field of stress and respiratory diseases such as asthma [37, 53, 57, 59], studies assessing the hypothalamic–pituitary–adrenocortical (HPA) axis [60, 61] are infrequent and none consider the autonomic response in early development (i.e., pregnancy, early childhood). Increasingly, evidence suggests that autonomic imbalance or dysfunction, independent of neuroendocrine or hormonal abnormalities [62–64] may be an understudied factor in the expression of a number of respiratory disorders. Indeed, research implicating autonomic imbalance in the pathogenesis of inflammatory and hypersensitivity reactions in the nose, skin, and the lung spans more than four decades [65–67]. This is because the ANS is integrally involved in regulation of airway function [68] and has important ties to immunoregulation. Notably, the HPA axis and ANS seem particularly susceptible to stress-induced programming.
Animal and human studies support the connection between an adverse intrauterine environment as well as experiences in early postnatal life and alterations of ANS functioning (e.g., sympathovagal balance) [69–72]. Animal research suggests that neural control of airway smooth muscle and irritant receptor systems is sensitive to environmental programming [69]. Respiratory and vagal systems undergo postnatal maturation to form an integration of respiratory and cardiovascular function [73, 74]. While our knowledge about the vulnerability of these systems to perinatal environmental influences and early programming is scarce [75], prenatal stress can increase allergen-induced airway inflammation [76, 77] and airway hyperresponsiveness (AHR) in prenatally stressed mice [78]. Others show exacerbations in airway inflammation in OVA-sensitized rats following repeated psychosocial challenge [79–83].
In humans, autonomic responses show developmental changes with relative stability between 6 and 12 months of age [84]. It seems reasonable that disruption of neuroendocrine and vagal anti-inflammatory pathways may predispose some individuals to immunodeviations and consequent disproportionate inflammatory responses resulting in altered respiratory responses. Balancing functional parasympathetic and sympathetic activity, in relation to emotional stimuli and immune function, may be central to understanding how psychological stressors influence airway inflammation and enhanced airway reactivity. Unfortunately, this has not been examined in human research. Nonetheless, reversible airway obstruction has been demonstrated during psychological challenge; cholinergic blockade supports a vagal origin [85, 86]. Negative affect in particular increases airway resistance [87] and it is suggested this occurs through vagal excitation to the airways [88]. Airway responses to induction of depressed mood are correlated with increased respiratory sinus arrhythmia (RSA) in asthmatics [89, 90]. Individuals with a propensity toward a greater vagal system response to distress may be prone to exaggerated airway narrowing. Interestingly, in an urban pregnancy cohort designed to study the effects of prenatal maternal and early life stress on urban childhood asthma risk, differential stress reactivity as indexed by prenatal HPA axis disruption [91] and cardiorespiratory parameters in infancy [92] has been observed.
Growing evidence also implicates a number of neurotrophins (NTs) as mediators or moderators of allergic disorders [93, 94] and shows that NT expression and signaling may be influenced by stress [95, 96]. One study in subjects with allergic asthma demonstrated that increased psychological stress was correlated with increased levels of brain-derived neurotrophic factor (BDNF) which, in turn, was negatively correlated with percent predicted forced expiratory volume in 1 s (FEV1) [97]. Notably, stress perception was also positively correlated with the percentage of TNF-alpha-producing T cells in these subjects. The authors suggest that this may indicate a neuroimmunological interaction given the ubiquitous secretion of BDNF in human peripheral blood monocytes which was enhanced when stimulated with TNF-alpha [98]. This group has also demonstrated stress-induced increase in tachykinin-like substance P associated with allergic airway inflammation in a mouse model [99].
Glucocorticoid resistance is an alternative hypothesis linking stress, neuroendocrine disruption (i.e., HPA axis), and immune function [100, 101]. Insight into the cellular and molecular mechanisms fundamental to stress-induced steroid resistance is provided in several studies. Oxidative stress pathways are likely involved in pathways linking psychosocial stress and respiratory diseases such as asthma [102] as well as steroid resistant asthma [103, 104]. This may be particularly applicable to airway inflammation where neutrophilic rather than eosinophilic inflammation predominates [105, 106]. Indeed, oxidative stress has been shown to contribute to steroid resistance in the context of neutrophilic inflammation in a mouse model of acute asthma exacerbations [107] supporting this hypothesis. While human data in the context of lung disease are sparse in this regard, one recent cross-sectional analysis in adolescents demonstrated that pBMCs harvested from asthmatics who perceived low parental support (i.e., greater stress) were more resistant to hydrocortisone’s effects on cytokine expression (IL-5, IFN-γ) and activation of eosinophils relative to asthmatics reporting higher parental support [108]. Further, glucocorticoid resistance disproportionately affects minority and low-income pregnant women [109] resulting in a limited ability to regulate inflammation. Examination of mechanisms contributing to steroid resistance in relation to increased environmental stressors may provide insight into the relationship between social stress, respiratory disease, and AHR across populations.
Oxidative Stress
While the mechanisms underlying environmental toxicities contributing to altered lung growth starting in utero are not fully elucidated, many lines of research point to a central role of oxidative stress and redox imbalance. Because reactive oxygen species (ROS) are thought to play a role in the etiology of disrupted lung growth and a number of respiratory disorders, in utero and early life exposures that promote (or diminish) the antioxidant defense system of the offspring may have lasting impacts. For example, studies show that factors that enhance antioxidant defense during pregnancy may have a persistent effect on antioxidant capacity that may mitigate oxidative stress-induced DNA damage later in life in tissues including the lung [110]. Social stress, like tobacco smoke exposure, is a pro-oxidant and may thus operate through these same pathways [102]. Parallel to research on intrauterine tobacco smoke exposure , epidemiological studies link increased prenatal stress with poor somatic growth [111–113]. This in turn is associated with smaller lungs and hence smaller airways, which also increase the risk of respiratory viral infections, altered lung function, wheeze, and other respiratory disorders [114, 115].
Emerging biomarkers of systemic oxidative stress include mitochondrial changes [116]. Mitochondria are major intracellular sources and primary targets of ROS, making them particularly susceptible to even small increases in systemic ROS [117]. Mitochondrial function can be altered by oxidative stress and inflammation [118]. In turn, dysfunctional mitochondria produce additional oxidation that may sustain systemic oxidative stress [118]. Alterations in mitochondrial function in the airway epithelium may intensify oxidative stress effects [119] suggesting mitochondrial-dependentpathways are important for airway remodeling and ultimately respiratory disease development [120]. Given that social stress has been shown to induce oxidative stress and damage [121], mitochondriomic changes may serve not only as a mechanistic function in the pathway from exposure to disease, but also as a “biomarker of stress.” Recent studies have implicated mitochondrial dysfunction in the etiology and symptomatology of many respiratory diseases [122–125]. Elevated serum lactate, an indicator of mitochondrial dysfunction [126], has been linked to both stable and acute severe asthmatic phenotypes [123, 124]. Variations in mitochondrial DNA have also been associated with elevated total serum IgE levels and augmenting Th2-type responses suggesting a role in the development of adaptive immunity [122, 127]. Mitochondrial changes have also been linked to the development and modulation of COPD, cystic fibrosis, and cancer [125]. Further, early life stress has been shown to induce mitochondrial changes via adenosine triphosphate (ATP) release and mitochondrial oxygen consumption [128, 129] suggesting alterations in these specific subcellular components likely play a role in stress-induced effects on the developing respiratory system and disease expression.
In addition to disparities in glucocorticoid resistance , high levels of oxidative stress, as measured by gamma-glutamyltransferase, a correlate of biologic exposure to pro-oxidant producing toxins [130], also disproportionately affect individuals of lower SES (education, occupational class, income). Similarly, increasing evidence suggests that minorities may be more likely to experience enhanced oxidative stress, oxidative DNA damage, and inflammation in response to environmental stimuli [131, 132]. These data suggest that the racial disparities and increasing prevalence of asthma , and other respiratory conditions, with decreasing SES may result from higher levels of oxidative stress among persons of lower relative to higher status and minorities.
Genetics and Epigenetics
Genetic factors of potential importance include those that influence immune development and airway inflammation in early life, corticosteroid regulatory genes, adrenergic system regulatory genes, biotransformation genes, and cytokine pathway genes. Variants of the glucocorticoid receptor gene have been shown to contribute to interindividual variability in HPA axis activity and glucocorticoid sensitivity in response to stress [133, 134]. Studies related to factors regulating the feedback mechanisms involved in the glucocorticoid response to stress are also of interest [135]. A recent study examined polymorphisms of the TNF-alpha promoter region (TNF-308G/A) and linked specific variants to increased C-reactive protein (CRP), a proinflammatory marker [136]. These are potentially interesting candidate genes to include in future studies of risk for atopic disease. Such studies that consider gene x environment interactions (i.e., stress by pathway genes) may inform specific mechanisms related to stress and atopy.
Programming effects of stress on respiratory outcomes may operate at a more fundamental molecular level, i.e., through epigenetic programming. Epigenetics may be at the roots of developmental plasticity imprinting environmental experiences on the fixed genome [137] albeit data are scare for respiratory health and allergic disorders [138, 139]. The epigenetic landscape has multiple layers, comprising of histone modifications, ncRNA, nucleosome positioning, and DNA methylation (the most studied epigenetic mechanism). In particular, DNA methylation is an adaptable epigenetic mechanism that modifies genome function through the addition of methyl groups to cytosine to form 5-methyl-cytosine (5mC). DNA methylation marks are largely established early in life [140] and may ensure stable regulation that mediates persistent changes in biological and behavioral phenotypes over the lifespan. Determining the range of environmental exposures that impact the epigenome during development was a research priority identified at the recent NHLBI Pediatric Pulmonary Disease Strategic Planning Workshop [141]. DNA methylation of many genes changes with disease status and in response to environmental signals including chemical exposures such as diet, drugs, and toxins. Recent findings also implicate psychological stress given behavioral studies demonstrating epigenetic changes during fear conditioning [142, 143] and evidence for epigenetic programming related to maternal care [144, 145].
The epigenome may be particularly sensitive to dysregulation in early development when DNA synthesis rates are highest. Genes involved in hypothalamic–pituitary–adrenal (HPA) axis functioning seem particularly susceptible to stress-related programming [146]. These include glucocorticoid receptor expression, the activation of which alters HPA activity through negative feedback inhibition. The human glucocorticoid receptor (GR) promoter region is extensively methylated with diverse methylation profiles demonstrated in normal donors [147]. The intracellular access of glucocorticoids to their receptors is also modulated by the 11 beta-hydroxysteroid dehydrogenase (11βHSD) enzymes , which interconvert biologically active 11 β-hydroxyglucocorticoids and inactive 11-ketosteroids [148]. While compromised 11βHSD2 activity can be caused by loss-of-function mutations of the gene encoding 11βHSD2, the frequency of such mutations is extremely low. Thus, other mechanisms accounting for the interindividual variability in 11βHSD2 enzyme activity should be considered. The 11βHSD2 promoter comprises a highly G + C-rich (or GC-rich) core, contains more than 80 % GC, lacks a TATA-like element, and has two typical CpG islands raising the possibility that methylation may play a role in the epigenetically determined interindividual variable expression of 11βHSD2.
Another candidate pathway implicated in both airway inflammation [149] and autonomic response [150] is the nitric oxide (NO) signaling pathways. Alterations of NO expression occur in the context of psychological stress and stress-related behaviors [151]. The inducible nitric oxide synthase (NOS) genes are also susceptible to epigenetic programming [152].
The notion that variability in methylation between subjects may reflect an important epigenetic mechanism is suggested by recent studies in both animals and humans. Epigenetic modulation of the 11βHSD2 gene has been recently demonstrated in a rodent model and cultured cell lines [153], albeit epigenetic regulation of this gene is not well characterized in humans. Weaver and colleagues have demonstrated differential methylation patterns of the NGFI-A-binding site in GR promoter 17 in the rat brain in offspring that had received poor maternal care versus those that had received better maternal care [154]. When pups were cross-fostered between dams providing good or poor postnatal care, the pups developed the epigenome of the foster mother. This same group reported increased methylation in a neuron-specific GC receptor (NR3C1) promoter as well as decreased levels of GC receptor mRNA from hippocampus tissue obtained from suicide victims with a history of childhood abuse [155]. Similar postnatal care has been linked to several hypermethylated regions upstream and downstream of the proximal GR promoter [156]. Recent human data demonstrates that methylation of exon 1 F in fetal cord blood was sensitive to maternal mood in the perinatal period and the infants HPA stress reactivity [157].
As highlighted earlier, the expression of neurotrophins, specifically BDNF, contributes to normal airway structure and function, and to airway hyperreactivity and remodeling in respiratory diseases. As reviewed by Prakash and Martin [158], sustained stress has been linked to hypermethylation of the BDNF gene , an effect which has been shown to begin in infancy and persist into adulthood, and consistently reduce BDNF mRNA and protein levels. Although early life adversity appears to influence the epigenetic markings of the BDNF gene, the underlying mechanisms are not completely clear.
In summary, genetic and epigenetic studies tell us that exposure to altered glucocorticoid receptor response through early development, even beginning in utero, programs major changes in the endogenous neuroendocrine and immune mechanisms that may, in turn, lead to increased vulnerability to respiratory disease. Whether alterations in DNA methylation underlie stress-induced phenotypic plasticity related to lung structure and function or disease risk remains largely unexplored in ethnically diverse populations. It will be important to begin to understand factors related to developmental programming of glucocorticoid sensitivity during critical periods of development which may play a role in disease etiology as well as subsequent morbidity.
Role of Stress in Respiratory Disease Morbidity: Asthma as an Evidence-Based Example
While asthma affects people of all ages, races, and ethnic groups, in the United States (U.S.), morbidity disproportionately burdens poor, ethnic minorities in both urban and nonurban environments [28, 29, 159]. Low SES has been consistently associated with greater asthma impairment, including more frequent emergency department visits [26] and greater symptoms/morbidity [27]. Similarly, racial/ethnic differences in asthma prevalence, asthma attacks, and increased emergency room visits for asthma exist among children and adults [160].
Asthma exacerbations are influenced by numerous factors including psychosocial influences [161, 162]. Trueba and Ritz [163] published an extensive review concluding individuals with existing asthma experiencing stress express a stronger bias toward Th2 activation . This is in line with the theory that stress effects Th cell functioning negatively increasing the risk of asthma exacerbations. The basic premise is that psychological stress can heighten airway inflammation in response to environmental triggers, and consequently increase the frequency, duration, and severity of an individual’s symptoms [164].
Low SES is consistently associated with greater asthma impairment, including more frequent emergency department visits, hospitalizations, greater symptoms, and more severe asthma [8, 165, 166], findings which are consistent regardless of the SES measure being explored. Ungar and colleagues [166] report that children with high income adequacy experience up to 28 % fewer exacerbations than children with low income adequacy and that every percentage increase in the proportion of income spent out of pocket on asthma medications was associated with a 14 % increase in exacerbations.
How does social stress help explain this observation? Poorer asthma morbidity may be greater among individuals of lower SES because they bear a disproportionate burden of exposure to suboptimal, unhealthy socially toxic environments. For example, in a study examining stressors immediately before or during pregnancy among a sample of 143,452 women [167], stress exposure increased as income decreased, with 57 % of low-income women experiencing at least one chronic stressor [e.g., economic hardship (37 %), job loss (19 %), separation or divorce (15 %), incarceration of partner (8 %), and domestic violence (5 %)]; 29 % experienced multiple stressors concurrently. In 2014, a study was published which explored the role of financial and social hardships in asthma racial disparities [28]. Compared to whites, African American caregivers experienced more social hardships including lower income and education attainment, difficulty finding work, having no one to borrow money from, not owning a car, and being single/never married. They reported that the SES and hardships explained 49 % of the observed racial disparity in asthma morbidity defined as hospital readmission. Effects of these stress exposures may be compounded among minorities by racism-related stressors.
Traumatic stressors may warrant particular consideration for many reasons [146]. Trauma, like other stress, occurs at increased rates among low-income, minority populations [168, 169]. Holman and colleagues [169] examined the rates of trauma in an ethnically diverse, community-based sample (N = 1456). Nearly 10 % experienced a trauma in the past year; 57 % reported at least one lifetime event including interpersonal violence occurring outside the family (21 %), acute losses or accidents (17 %), witnessing death or violence (13 %), and domestic violence (12 %). Hien and Bukszpan [170] examined lifetime interpersonal violence among a “control” group of urban, low-income women, predominantly Latina or blacks, who had been screened for the absence of psychopathology. Almost 28 % of these urban women reported a history of childhood abuse, compared to general population estimates of 10 %. Urban minority women also experience heightened levels of community violence [171, 172]. Other studies have documented increased rates of PTSD and depression in urban samples [146]. The perinatal period is a vulnerable time to experience more intense psychological symptoms, particularly for low-income women. Compared to other forms of stress, trauma is more likely to result in psychological morbidity [e.g., posttraumatic stress disorder (PTSD) , depression] and persistent psychophysiological changes (HPA axis, sympathetic-adrenal-medullary [SAM] system). One study in urban children found a relationship between exposure to chronic str ess (violence) in early childhood and reduced lung function at age 6 years [173] suggesting these effects persist years after exposure, particularly when the exposure occurs during a critical developmental window (e.g., when stress regulatory systems are developing).
Need for a Multilevel Framework
The etiology of respiratory health problems is increasingly recognized as a result of the complex interplay of influences operating at several levels, including the individual, the family, and the community (Fig. 8.2). Ecological views on health recognize that individual-level health risks and behaviors have multilevel determinants, in part influenced by the social context within which subjects live [174]. That is, chronic stress experiences are significantly influenced by the characteristics of the families, homes, and communities in which we live [175, 176]. Both physical and social factors can be a source of environmental demands that contribute to stress experienced by populations living in a particular area [177].
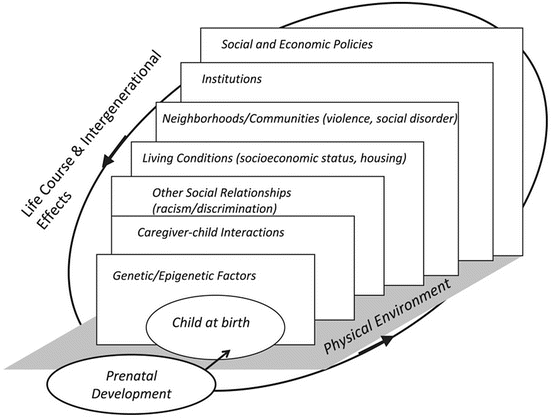
Fig. 8.2
Characterizing stress across the life course: an ecological approach
Taking a multilevel approach to examining stress effects on respiratory disease development , including asthma, may be particularly relevant to the understanding of disparities based on race/ethnicity and SES [176]. This includes an environmental justice perspective underscoring the role of structural and macrosocial forces that shape exposure and vulnerability to diseases may better inform the complex social patterning of asthma [176]. According to this framework, asthma rates are higher and the associated morbidity is greater among the poor because they bear a disproportionate burden of exposure to suboptimal, unhealthy environmental conditions. Upstream social and economic factors determine differential exposures to relevant asthma pathogens and toxicants [16]. Also, understanding the upstream factors (e.g., social and economic policies) that contribute to the varying social conditions for populations and individuals being studied will better inform needed interventions.
Life Course Perspective and Intergenerational Effects
Other studies provide evidence supporting the intergenerational transmission of psychophysiological vulnerability in traumatized populations. While studies of maternal stress and infant outcomes typically examine events occurring during pregnancy, we recently considered stress (interpersonal trauma, IPT) across the mother’s life course in relation to early immune markers in their children [178]. The life course perspective posits that some stressors may influence health through two mechanisms, early programming and cumulative pathways, in addition to more immediate effects. Early programming may occur if exposures during sensitive developmental periods in the mother have lasting psychobiologic sequelae. Exposure to IPT in earlier life can generate disrupted physiological stress responses even several years following the trauma. Thus, maternal IPT may be linked to infant health through more latent effects (i.e., lasting effects from abuse in childhood/adolescence), proximate effects (i.e., trauma experienced in or around the pregnancy), and cumulative life course effects (i.e., allostatic load of accumulated traumas over the mother’s life). It has been demonstrated that infants born to mothers with chronic trauma exposure—that is, both early in life and more proximate to the pregnancy—would be at greatest risk of expressing elevated IgE [178].
Constricting Communities
Indicators of neighborhood disadvantage, characterized by the presence of a number of area-level stressors including poverty, unemployment/underemployment, percentage of unskilled laborers, limited social capital or social cohesion, substandard housing, and high crime/violence exposure rates, have been investigated in relation to urban children’s development [176]. Such stress is chronic and can affect all subjects in a given environment regardless of their individual-level risks.
Growing evidence suggests that community violence, a risk factor experienced disproportionately by lower SES individuals, may contribute to the burden of asthmain urban populations . This notion is supported by three longitudinal studies. Sternthal et al. [179] analyzed over 2000 urban children in Chicago between the ages of 0 and 9 and reported a significant association between higher community violence exposure and increased risk for asthma development in urban children. This association was robust after controlling for important individual-level factors (race/ethnicity, SES, maternal health behaviors, family violence) and neighborhood-level confounders (concentrated disadvantage, social disorder, and collective efficacy). In an ethnic minority population made up of mainly low SES participants, Chiu and colleagues [180] observed independent effects of prenatal community violence exposure and physical toxins at the neighborhood level (e.g., air pollution) on repeated wheeze. Similarly, lifetime community violence exposure among ethnically diverse 6- to 7-year-olds has been linked to poorer lung function (e.g., lower FEV1) [173]. In addition to asthma development and lung function detriments, increased exposure is also associated with higher hospitalization rates and emergency department visits [181].
Housing Stressors
A number of subjective housing characteristics have been linked to adverse psychological outcomes. This subjective emotional dimension of housing may influence asthma outcomes [8, 182] although this is only starting to be empirically explored. Specifically, housing stress related to lack of utilities, furniture, appliances, and relationships with neighbors/landlords has been linked to poor asthma control, school absences, unplanned visits to the emergency department, and exercise intolerance [183]. Further, housing disarray, characterized by a dark, cluttered, crowded, or noisy house, has been associated with increased asthma prevalence among children [184]. Studies have shown that improved housing can have a positive impact on asthma severity including decreases in lost school/work days, disturbed sleep, and asthma symptoms [185]. Improving housing should be a focus for any intervention efforts.
Family Factors
There have been a number of examples from the asthma epidemiology literature showing associations between early caregiver stress and the development of asthmatic phenotypes in early childhood [186, 187]. Suglia and colleagues [188] recently demonstrated that maternal ability to maintain positive caregiving processes in the context of even more extreme stress may buffer the effects on child asthma risk. They examined the prospective relationship between maternal intimate partner violence (IPV) and asthma onset in children in the Fragile Families and Child Wellbeing Study (N = 3117), a birth cohort. Maternal report of IPV was assessed after the child’s birth and at 12 and 36 months. Mothers also indicated how many days a week they participated in activities with the child and the amount and type of educational/recreational toys available for the child. Maternal report of physician-diagnosed asthma by age 36 months was the outcome. In adjusted analysis, children of mothers experiencing IPV chronically (at all time periods), compared to those not exposed, had a twofold increased risk of developing asthma. In stratified analysis, children of mothers experiencing IPV and low levels of mother–child activities (RR 2.7, 95 % CI 1.6, 4.7) had a significant increased risk for asthma. Those exposed to IPV and high levels of mother–child activities had a lower risk for asthma (RR 1.6, 95 % CI 0.9, 3.2). One should also consider the developmental timing of exposures over the life course relative to specific asthma outcomes whether individual or contextual factors are being considered. Factors leading to the onset, remission, or persistence of asthma across the life course may be influenced by social experiences and physical exposures beginning in utero, a series of social and biologic experiences initiated by early childhood exposure or cumulative exposure to toxic biologic or social factors over critical periods of development. It is important to consider stress at these multiple levels given that they are interrelated throughout the life course. If we can understand at what level stress is occurring and perhaps has the greatest impact on asthma expression, this may inform the most effective interventions.
Stress-Enhancing Effects on Physical Environmental Exposures
Because of the covariance across exposures and evidence that social stress and other environmental toxins (e.g., pollutants, tobacco smoke) may influence common physiological pathways (e.g., oxidative stress, pro-inflammatory immune pathways, autonomic disruption), understanding the potential synergistic effects promises to more completely inform children’s asthma risk [15]. Epidemiological studies have demonstrated synergistic effects of stress and air pollution on asthma expression among children and adolescents [180, 189–191]. We need to better understand how the physical and psychological demands of living in a relatively deprived environment may potentiate an individual’s susceptibility to cumulative exposures across these domains.
Summary
Social toxicity experienced as increased psychological stress is likely a major driver of observed disparities in lung growth and development and asthma, as well as a range of other respiratory conditions. Most respiratory conditions likely share overlapping etiology; therefore, multiple mechanistic pathways with complex interdependencies must be considered when examining the integrative influence of stress independently as well as the interaction of social and physical environmental toxins in explaining the social patterning of respiratory diseases. Because these factors tend to cluster in the most socially disadvantaged, this line of research may better inform the etiology of growing health disparities increasingly documented for respiratory disorders. Future epidemiologic studies which concomitantly consider social stress as well as physical environmental toxins will likely more fully explain the etiology of respiratory disease disparities as well as inform more effective prevention and intervention strategies that enhance lung growth and reduce respiratory disease morbidity.
Acknowledgements
During preparation of this chapter KJ Brunst was supported by the National Institutes of Health, K99ES024116.
References
1.
Castro-Diehl C, Diez Roux AV, Seeman T, Shea S, Shrager S, Tadros S. Associations of socioeconomic and psychosocial factors with urinary measures of cortisol and catecholamines in the Multi-Ethnic Study of Atherosclerosis (MESA). Psychoneuroendocrinology. 2014;41:132–41.PubMedCentralPubMed
2.
Agardh E, Allebeck P, Hallqvist J, Moradi T, Sidorchuk A. Type 2 diabetes incidence and socio-economic position: a systematic review and meta-analysis. Int J Epidemiol. 2011;40(3):804–18.PubMed
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


