(1)
Project-team INRIA-UPMC-CNRS REO Laboratoire Jacques-Louis Lions, CNRS UMR 7598, Université Pierre et Marie Curie, Place Jussieu 4, 75252 Paris Cedex 05, France
Abstract
Smooth muscle cells, or smooth myocytes (SMC), similarly to any myocyte, contract and relax. Yet, unlike striated myocytes, they can withstand prolonged stretch. Smooth myocytes can contract phasically with rapid contraction and relaxation such as in the digestive tract, where they create peristaltism. Smooth myocytes can also contract tonically with slow and sustained contraction, such as in the cardiovascular and respiratory apparatus.
Smooth muscle cells, or smooth myocytes (SMC), similarly to any myocyte, contract and relax. Yet, unlike striated myocytes, they can withstand prolonged stretch. Smooth myocytes can contract phasically with rapid contraction and relaxation such as in the digestive tract, where they create peristaltism. Smooth myocytes can also contract tonically with slow and sustained contraction, such as in the cardiovascular and respiratory apparatus.
Transient and sustained contraction of smooth myocytes relies on 2 different pathways. (1) Transient, calmodulin-dependent contraction results from activation of G-protein-coupled receptors by their specific ligands, such as substance-P and cholecystokinin, that trigger production of inositol (1,4,5)-trisphosphate and calcium influx in the SMC cytosol. (2) Prolonged contraction is also caused by chemical messengers, such as bombesin and ceramide, that prime extracellular calcium influx and a protein kinase-C-dependent, calmodulin-independent axis.
Smooth myocytes pertain to involuntary (i.e., without conscious control), non-striated myocytes. They control the dimension and mechanical function of hollow organs and anatomical conduits. In particular, smooth myocytes lodge in walls of both the vasculature (arteries, veins, and large lymphatic vessels) and large and mid-sized segments of the ventilatory tract that experience large deformations during the cardiac and respiratory cycles. In both structures, they reside beneath the cell layer that lines the conduit lumen, i.e., the vascular endothelium and respiratory epithelium, respectively.
Smooth myocytes are innervated by theautonomic nervous system.1 According to the unitary or collective innervation of SMC sheets or bundles, smooth myocytes constitute 2 groups. (1) In single-unit smooth muscles, the autonomic nervous system innervates a single cell within the SMC set. The command electrochemical wave, oraction potential, propagates through gap junctions to apposed cells, such that the entire cell set contracts in bulk, as a syncytium. Single-unit smooth muscles line blood vessels, except large elastic arteries. (2) In multi-unit smooth muscles, such as in the trachealis muscle that connects the ends of U-shaped cartilaginous rings of the trachea and media of large arteries, the autonomic nervous system innervates many cells to finely tune nervous control and cause gradual responses.
Smooth myocytes can contribute to the formation of theextracellular matrix, as they secretecollagens, mostly type-1 and -3,elastin,glycoproteins, andproteoglycans. They possess on their surface elastin and collagen receptors that interact with these extracellular proteins.
The plasma membrane, or sarcolemma, possesses specialized nanodomains devoted to cell signaling with ion carriers and receptors for hormones, growth factors, neurotransmitters (e.g., acetylcholine and cathecholamines), and regulators of the SMC tone (e.g., nitric oxide and endothelin). However, smooth myocytes in various regions of the vascular bed, airways, and kidneys differ in their production of ion carriers and receptors, as well as other proteins that determine their function.
8.1 Markers of Smooth Myocytes
Smooth myocytes of walls of the vasculature and ventilatory tract possess various contractile proteins. Contractility of smooth myocytes relies on smooth muscleα-actin,myosin heavy chain,calponin,2caldesmonhMW,3.β-tropomyosin, andmyosin light chain kinase (MLCK).
Hypercontractility markers include myosin light chain kinase, smooth muscle-specific myosin heavy chain (smMHC) isoforms, and 22-kDa actin-binding, smooth muscle protein (SM22), or transgelin (TaGln). In addition to cytoskeleton constitutents, calmodulin is synthesized in smooth myocytes to participate in the control of cytoskeleton functioning.
Myosin-2 and actin generate cell contraction viastress fibers. In contracting smooth myocytes, β-actin network is conserved, whereas α-actin and myosin-2 filaments remodel. Myosin-2 is activated by Ca2 + –calmodulin myosin light chain kinase and inhibited bymyosin light chain phosphatase.
Contraction of smooth myocytes is regulated not only by phosphorylation of the myosin light chain by Ca2 + –calmodulin-dependent myosin light chain kinase, but also by the caldesmon–calmodulin complex,protein kinase-C, and calponin.
8.1.1 Actin
Two classes and 3 main groups ofactin isoforms exist: class-1, non-muscle actin, which encompasses β and γ groups, and class-2, muscle actin, which corresponds to α group. In myocytes, α actins are constituents of the contractile apparatus.
Smooth myocytes possess class-2 α2- and α3- (or γ2)-actins, in addition to class-1 actins. Isoforms β and γ coexist in most cell types as components of microfilaments of the cytoskeleton that support signal transduction, cell adaptation to mechanical loads, intracellular transport, and cell motility, among other functions (Vol. 1 – Chap. 6. Cell Cytoskeleton).
8.1.2 Tropomyosin
Tropomyosin polymers (TpM1–TpM4) are actin-binding proteins that regulate the binding of myosin, as they block access to myosin-binding sites on actin. Tropomyosins reside along each of the 2 grooves of actin filaments to yield structural stability and modulate filament function.
Many isoforms are produced by alternative promoters of 4 genes (TPM1–TPM4) as well as RNA processing. Many of these isoforms have a tissue- and filament-specific distribution.
8.1.3 Myosin
Myosin are adenosine triphosphatases that constitute a superfamily (Vol. 1 – Chap. 6. Cell Cytoskeleton). Non-conventional myosins, such as myosin-5 and -6 that travel toward the barbed (or + ) and pointed (or − ) ends of actin filaments, respectively, are devoted to cargo transfer within the cell. Conventional myosin, i.e., myosin-2, contains 2 heavy and 2 pairs of light chains. Myosin heavy chains contain N-terminal heads and C-terminal tails that hold the 2 heavy chains together in a coiled-coil manner. Two myosin regulatory light chains per heavy chain N-terminus bind the region (neck) between the head and tail.
Four known isoforms of smooth muscle myosin heavy chain (smMHC) are formed by 2 independent, alternative splices of the transcript of a single gene (smMHC1a–smMHC1b and smMHC2a–smMHC2b).4 Isoforms of the mMHCib type contain an additional 7 amino acids that double MgATPase activity as well as the displacement rate on actin filaments [705].
The ratio between actin and myosin is greater in smooth myocytes (15/1) than striated myocytes (7/1). This ratio is very large in non-myocytes. α-Smooth muscle actin (α smA), localized in microfilament bundles, is typical of smooth myocytes.
8.1.3.1 Myosin Light Chain Kinase and Phosphatase
The myosin regulatory system is more important in smooth muscles than striated muscles. The myosin light chain (MLC) phosphorylation state that determines actin polymerization associated with smooth muscle contraction is tightly controlled by the relative activities of antagonist enzymes myosin light chain kinase(MLCK) and phosphatase(MLCP; Fig. 8.1).
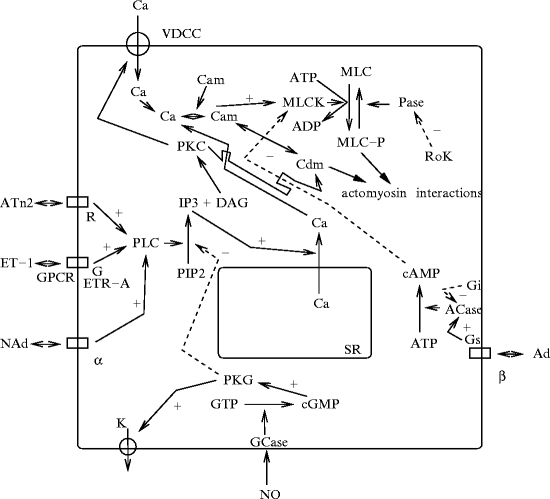
Fig. 8.1
The smooth myocyte function and its regulation (Source: [761]). Two pathways are involved: (1) the phosphatidylinositol and (2) Gs-protein axes. A ligand activates a GPCR, which stimulates phospholipase-C (PLC), producing inositol trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol bisphosphate (PIP2). IP3 releases calcium from the sarcoplasmic reticulum (SR) and DAG activates protein kinase-C (PKC). PKC acts on the contractile proteins and ion channels. Calcium and calmodulin (Cam) stimulate a myosin light chain kinase (MLCK), leading to actin–myosin interactions. Noradrenaline (NAd), acting via α1-adrenoceptors, angiotensin-2 (ATn2) via its cognate receptors, and endothelin-1 (ET1) via ETRa receptors (ETA; in fugure ETR-A) activate phospholipase-C (PLC). Gs protein activated by a β2-adrenoceptor agonist stimulatesadenylate cyclase (ACase), which catalyzes the formation of cAMP messenger. The latter inhibits MLCK and causes vasodilation.
Myosin light chain kinase activated by calcium–calmodulin increases sensitivity of myosin to calcium and regulates actin–myosin interactions. Phosphorylation of MLC leads to cross-bridge formation between myosin heads and actin filaments, and hence to smooth muscle contraction. Dephosphorylation of myosin light chains by myosin light chain phosphatase causes smooth muscle relaxation.
8.1.3.2 Small RhoA GTPase and RoCK Kinase
The RhoA–RoCK pathway inhibits myosin phosphatase. Stimulation of GPCRs generates smooth myocyte contraction via Ca2 + –calmodulin-dependent activation of MLCK and subsequent myosin phosphorylation. Kinase RoCK also regulates MLC phosphorylation. Myosin phosphatase- and RhoA-interacting protein (MPRIP)5 is detected withprotein kinase-G and myosin phosphatase and colocalize with myofibrils in smooth myocytes.
8.1.4 Calmodulin
Calmodulin (i.e., calcium-modulated protein) is an ubiquitous, multifunctional regulator and calcium sensor. It controls numerous cell functions, such as metabolism (calcium, cyclic nucleotide, and glycogen), secretion, smooth muscle contraction, intracellular transport, and gene expression.
Calmodulin isoforms (Cam1–Cam3) are encoded by 3 genes in humans. In addition to these isoforms, the calmodulin family contains calmodulin-like proteins (CalmL1 and CalmL3–CalmL6)6
8.1.5 Caldesmon
Binding partners ofcaldesmon include actin, Ca2 + –calmodulin, myosin, tropomyosin, and phospholipids. Caldesmon also binds caltropin [706] and tubulin [707]. Alternatively spliced transcript variants generate diverse isoforms.
Caldesmon is implicated in the regulation of actomyosin interactions in smooth myocytes as well as non-myocytes (Table 8.1). Caldesmon precludes the ATPase activity of myosin in smooth myocytes, in addition to the regulation of non-myocyte contraction.
Table 8.1
Regulatory proteins of contraction. Caldesmon can be sequestered away from actin by calcium–calmodulin complexes; it is phosphorylated by PKC.
CMC | SMC |
|---|---|
Troponin | Caldesmon |
Caldesmon reversibly binds to Factin and actin–tropomyosin filaments. In particular, the interaction of high-molecular-weight caldesmonhMW with actin–tropomyosin filaments hindersmyosin ATPase activity. Calmodulin binds to caldesmonhMW at activating calcium concentrations, thereby hampering the interaction of caldesmonhMW with actin–tropomyosin to activate the myosin ATPase activity and cause contraction.
Mitogen-activated protein kinase that is regulated byprotein kinase-C phosphorylates caldesmon, especially under smooth myocyte stretching [708].
8.1.6 Calponin
Calponin is a calcium- and calmodulin-binding protein that prevents the MgATPase activity of myosin in smooth myocytes [709]. Calponin also connect to tropomyosin, independently of Ca2 + , but links to calmodulin in the presence of Ca2 + ion (Table 8.2). Calponin is phosphorylated byprotein kinase-C andCa2 + –calmodulin-dependent protein kinase CamK2 to relieve calponin inhibition of myosin.
Table 8.2
Calponin-binding proteins (Source: [710]). Addition of calponin to actin–tropomyosin complex provokes formation of actin bundles.
Partner | Function |
|---|---|
Actin | Modulation of actin–myosin-2 ATPase activity |
α-Actinin | Stabilization of the α-actinin–actin complex |
Caldesmon | Myosin-2 binding regulation |
Caltropin | Weakening of caldesmon–actin interaction |
Calmodulin | Inhibition of actin binding |
Myosin-2 | Stimulation of myosin-2 ATPase activity |
Tropomyosin | Actin stability and myosin-2 regulation |
Tubulin | Microtubule binding |
HSP90 | Prevention of actin bundle formation |
Decrease in calponin-induced polymerization of Gactin | |
CamK2 | Phosphorylated |
ERK1/2 | Scaffolding for ERK signaling |
PKC | Phosphorylated |
8.2 Glycocalyx
Heparan sulfate proteoglycans of theglycocalyx (thickness 5–11 μ [711]), such as syndecan-1 and -4, can cooperate with integrins to launch cell spreading and assembly of focal adhesions and stress fibers.
Interstitial flow upregulatesmatrix metalloproteinase expression in rat vascular smooth myocytes, fibroblasts, and myofibroblasts via theFAK–ERK1/2-Jun pathway [712]. At the surface glycocalyx of rat vascular smooth myocytes,heparan sulfate proteoglycans sense interstitial flow andtransduce this mechanical signal. Subsequent phosphorylated (activated) of mechanosensitive focal adhesion kinase enable ERK1/2 activation, interstitial collagenase (MMP13) expression, and smooth myocyte motility.
8.3 Intrinsic Regulators of Smooth Myocyte Tone
8.3.1 Caldesmon and Calmodulin
Caldesmon may act on Factin by either phosphorylation byprotein kinase-C andCa2 + or formation of a caldesmon–calcium–calmodulin complex that is unable to bind to actin. Moreover, caldesmon enhances the binding of ATP-bound heavy meromyosin (HMM), as well as (but to a lesser extent) the subfragment S1–ATP (like HMM–ATP in skeletal myocytes), to actin [713]. This effect is hindered by Ca2 + –calmodulin. Caldesmon also interacts with myosin in a process regulated bycasein kinase-2 [714].
8.3.2 Arachidonic Acid
Arachidonic acid stimulates ATPase activity of unphosphorylated smooth muscle myosin, heavy meromyosin, and subfragment S1, thereby causing contraction of smooth myocytes [715].
8.3.3 G-Protein-Coupled Receptors
In smooth myocytes, signaling fromG-protein-coupled receptors controls contraction and growth as well as tissue remodeling and inflammation. Gq-coupled receptors operate viaphospholipase-Cβ that hydrolyzes phosphatidylinositol (4,5)-bisphosphate into intracellular messengersdiacylglycerol andinositol (1,4,5)-trisphosphate.
Cytosolic Ca2 + concentration increases upon the release from sarcoplasmic reticulum stores through IP3 receptors and influx from plasmalemmal IP3 channels (Vol. 3 – Chap. 3. Main Classes of Ion Channels and Pumps). Elevated intracellular calcium level causes Ca2 + binding to calmodulin. Calcium–calmodulin then activatesmyosin light chain kinase. The latter phosphorylates myosin light chains to enable actin to activate myosin ATPase activity for contraction of actomyosin filaments.
Diacylglycerol that remains bound to the plasma membrane initiates the translocation ofprotein kinase-C from the cytoplasm to the membrane and its subsequent activation. Protein kinase-C phosphorylatescalponin to relieve its inhibition of myosin ATPase. It also phosphorylates components ofMAPK modules (Vol. 4 – Chap. 6. Mitogen-Activated Protein Kinase Modules) that activate transcription factors involved in cell growth. In addition, PKC stimulatesphospholipase-A2 that releases arachadonic acid.
Gq-coupled receptors also interact with guanine nucleotide-exchange factors of Rho GTPases (RhoGEFs) to regulate actin network reorganization. MonomericRho GTPase activatesRoCK kinase that phosphorylates the myosin-binding subunit ofmyosin light chain phosphatase to inhibit its activity, thereby heightening phosphorylation of myosin light chain.
8.3.4 Guanylate Cyclases, Nitric Oxide, and Natriuretic Peptides
Nitric oxide andatrial natriuretic peptide relax smooth myocytes viasoluble andparticulate guanylate cyclases, cGMP messenger andcGMP-dependent protein kinase isozymes PKG1α and PKG1β.7 Both isoforms have several common substrates, such as inositol (1,4,5)-trisphosphate receptor (IP3R)-associated cGMP kinase substrate (IRAG),8 myosin-binding PP1r12a regulatory subunit, andregulator RGS2 of G-protein signaling.
Enzyme PKG1β phosphorylates PP1r12a, hence causing calcium desensitization of myosin regulatory light chain. It also targets RGS2, thereby modulating receptor-mediated signaling via Gq/11 class of G-protein subunits. It interacts specifically with IRAG protein that is highly expressed in the membrane of the endoplasmic reticulum of smooth myocytes. It allows stable interaction of IP3R1 receptor with PKG1β enzyme [716].
8.3.5 Inositol Trisphosphate Receptors and Calcium Release
Entry of extracellular calcium can occur through plasma membrane ion carriers, such asvoltage-dependent CaV1.2 channels, non-selective cation channels, andNa + –Ca2 + exchangers. Intracellular calcium signals are also produced by Ca2 + 2+ release from intracellular stores (sarco[endo]plasmic reticulum and mitochondria).
In smooth myocytes, calcium signals include Ca2 + flashes,9 oscillations,10 puffs,11 ripples,12 sparklets, sparks,13 waves,14 and global intracellular Ca2 + concentration elevation (Table 8.3).15 Activated IP3R generates these cues. However, non-selective cation, store-operated Ca2 + , ryanodine-sensitive Ca2 + release, transient receptor potential, and voltage-dependent Ca2 + channels also contribute to and modulate these calcium signals.
Table 8.3
Calcium signals regulated directly or indirectly by IP3Rs in smooth myocytes (Source: [717]; CaV: voltage-dependent Ca2 + channel; NEx: Na + –Ca2 + exchanger; NSCC: non-selective cation channel; RyR: ryanodine-sensitive Ca2 + release channel; SOC: store-operated Ca2 + channel; TRPC: transient receptor potential canonical channel; TRPM: melastatin-related transient receptor potential channel).
Calcium | Frequency | Rise | Decay | Spatial | Involved |
|---|---|---|---|---|---|
signals | (Hz) | time | half-time | spread | Ca2 + |
(s) | (s) | (μ m) | channels | ||
Flash | 0.2–0.5 | 0.2–0.8 | 0.5–1.5 | CaV, RyR | |
Global | CaV, NSCC, | ||||
spread | RyR, SOC | ||||
TRPC1/3, | |||||
TRPM4 | |||||
Oscillation | 0.07–0.4 | 2–6 | 3–18 | RyR, | |
TRPC6 | |||||
Puff | 0.02–0.04 | 0.06–0.16 | 0.11–0.25 | 1.89–2.51 | RyR |
Ripple | 0.05–0.19 | 1–2 | 4–6 | ||
Spark | 0.07–0.59 | 0.01–0.07 | 0.03–0.15 | 0.52–4.7 | RyR |
Wave | 0.04–1.41 | 0.5–2 | 2–10 | 8.8–50 | CaV, |
NSCC, NEx, | |||||
RyR, SOC |
Plasmalemmal Gq/11-coupled receptors stimulatephospholipase-C that hydrolyzes plasmalemmal phosphatidylinositol (4,5)-bisphosphate and generates diacylglycerol and inositol (1,4,5)-trisphosphate.
Inositol trisphosphate receptors constitute a set of tetrameric intracellular Ca2 + release channels on the endoplasmic reticulum membrane that generate Ca2 + signals (Vol. 3 – Chap. 3. Main Classes of Ion Channels and Pumps). Three IP3R isoforms (IP3R1–IP3R3; Table 8.4) encoded by distinct genes are responsible for signaling differences with given frequencies, amplitudes, and spatiotemporal properties. These 3 isoforms are synthesized in smooth myocytes. Subtype IP3R1 is the predominant isoform in vascular smooth myocytes; IP3R2 and IP3R3 densities are higher in proliferating vascular smooth myocytes [717]. In the tracheal smooth muscle, IP3R3 is the principal isoform, IP3R1 and IP3R2 being also expressed [717].
Table 8.4
Main features of IP3R isoforms (Source: [717]). In vascular smooth myocytes, IP3R receptors localize to the central sarco(endo)plasmic reticulum around the nucleus as well as its peripheral region beneath the plasma membrane. They thus can regulate Ca2 + -dependent gene expression as well as local signaling to membrane proteins. IP3R channel is activated at a cytosolic Ca2 + level < 300 nmol; an elevation in cytosolic Ca2 + level together with calmodulin inhibit IP3R channel. Agent ATP is not required for IP3R activation, but enhances sensitivity to IP3 and Ca2 + ligands. Binding of IP3 to IP3R in aortic and tracheal smooth myocytes is weak at pH < 7 and maximal at pH ranging from 8 to 9. In portal vein smooth myocytes, a pH increase from 6.7 to 7.0–7.3 reduces the Ca2 + concentration required for IP3R activation. Reactive oxygen species stimulate IP3R-mediated Ca2 + release in aorta smooth myocytes, whereas they impede SERCA pump in coronary artery smooth myocytes, but do not affect IP3R activity in mesenteric artery smooth myocytes.
IP3R1 | IP3R2 | IP3R3 | |
|---|---|---|---|
IP3 affinity | Intermediate | Highest | Lowest |
( ∼ 270 nmol) | ( ∼ 100 nmol) | ( ∼ 400 nmol) | |
Activation by Ca2 + | ∼ 170 nmol | ∼ 150 nmol | ∼ 60 nmol |
Inhibition by Ca2 + | ∼ 370 nmol | ∼ 160 nmol | ∼ 170 nmol |
Sensitivity to ATP | High | None | Low |
( ∼ 130 μ mol) | ( ∼ 2 mmol) | ||
Influence of pH | 6.8 | 7.5 | |
(IP3 binding, | |||
Ca2 + sensitivity) | |||
Alternative splicing | Yes | None described | None described |
The cytosolic N-terminus possesses an IP3-binding domain, a suppressor motif that inhibits IP3 binding, and a regulatory sequence with binding sites for ATP and Ca2 + ion as well as phosphorylation sites and a coupling region for interacting with transient receptor potential canonical (TRPC) channels [717]. The transmembrane region and cytosolic C-terminus enable IP3R tetramerization.
Density of IP3Rs results from the balance between gene transcription and protein degradation.Transcription factor MyB binds to the Ip3r1 promoter and stimulates transcription, whereas retinoic acid precludes it viaActivator protein-2 [717].Hydrogen peroxide promotes proteasomal degradation of IP3R1 and IP3R3, whereasJaK2 kinase phosphorylates IP3R1 and prevents its proteasomal degradation.Vasopressin impedes IP3R1 gene transcription and favors protein degradation [717].
Several ligands (IP3, cytosolic and sarco(endo)plasmic reticulum luminal Ca2 + , and ATP), kinases (inhibitory PKA and PKG and stimulatory PKC and Tyr kinases), and other types of regulators (FKBP12 via inhibitory PP3 and stimulatory TOR, scaffold enhancer RACK1) and modulators (cellular pH16 and reactive oxygen species) control IP3R activity [717].
In addition, IP3R communicates with adjoiningryanodine-sensitive Ca2 + channels of the sarco(endo)plasmic reticulum as well as mitochondria to influence Ca2 + release from its main store as well as reactive oxygen species generation. It can couple to plasmalemmaltransient receptor potential channels (e.g., TRPC1 and TRPC3),17store-operated Ca2 + channels,18 andlarge-conductance Ca2 + -activated K + channels (BK),19 which lodge incaveolae where the sarco(endo)plasmic reticulum and plasma membrane are in close proximity [717].
In airway and vascular smooth myocytes, proximity (distance ∼ 30 nm) betweenmitochondria and sarco(endo)plasmic reticulum enables local signaling between these 2 organelles. In particular, IP3R regulates mitochondrial Ca2 + concentration.Mitochondrial Ca2 + uniporter, the major mitochondrial Ca2 + uptake axis, is sensitive to micromolar Ca2 + levels. IP3R-mediated Ca2 + waves increase mitochondrial Ca2 + concentration, stimulate ROS generation and NFκ B, and foster CaV1.2 gene expression in cerebral artery smooth myocytes [717].
Conversely, mitochondria regulate IP3R in smooth myocytes, as they: (1) buffer local and global elevation in cytosolic Ca2 + level; (2) control ATP synthesis;20 (3) participate in ROS signaling;21 and modulate ion channels that regulate membrane potential and cytosolic Ca2 + level. In vascular smooth myocytes, mitochondria regulate the activity of several plasmalemmal ion channels, such as voltage-dependent Ca2 + , Ca2 + -activated K + , Ca2 + -activated Cl − (ClCa), and store-operated Ca2 + channels, thereby influencing membrane potential and Ca2 + signals [717].
Inositol trisphosphate receptors controls not only cell contraction, but also gene expression and cell migration and proliferation. In cerebral artery smooth myocytes, IP3R-mediated Ca2 + release activates NFκ B directly and indirectly via mitochondrial ROS production; NFκ B stimulates CaV1.2 channel α1c subunit expression [717]. Cell proliferation correlates with an elevation in IP3R1 production in aortic and carotid artery smooth myocytes and upregulation of all IP3R isoforms in mesenteric artery smooth myocytes [717].
8.4 Airway Smooth Myocytes
8.4.1 Mechanical Loading-Induced Cytoskeleton Remodeling
Airway smooth myocyte (aSMC) is able to modify its shape, stiffness, and contractility in response to mechanical loading after adaptation of actomyosin interaction andintegrins at adhesion junctions [718].
In the tracheal muscle, strains elevate phosphorylation level of bothfocal adhesion kinase andpaxillin. Once phosphorylated by focal adhesion kinase, adhesion complex component paxillin couples toneuronal Wiskott-Aldrich syndrome protein (nWASP) viaCRK adaptor to launch nWASP activation by activatedCDC42 GTPase that binds to recruited nWASP (Vol. 1 – Chap. 6. Cell Cytoskeleton and 7. Plasma Membrane). The latter stimulates theARP2–ARP3 complex that polymerizesactin filaments.
Integrin-linked kinase and its binding partners coordinate cytoskeleton remodeling, especially formation of structural links between integrins and the actin cytoskeleton. Integrin-linked kinase tethers to β1-integrins and can then transduce signals from integrin receptors to regulate actin polymerization. This scaffold forms a heterotrimer withLIMS1 adaptor (or PINCH) andα-parvin that binds to actin filaments. Enzyme ILK also connects to paxillin. In addition, LIMS1 adaptor may couple to nWASP viaNCK adaptor.
8.4.2 Contractile and Proliferative Phenotype
Airway smooth myocyte can evolve from a contractile to a proliferative phenotype. When aSMCs proliferate, the content in smooth muscle α-actin, high-molecular-weight caldesmonhMW isoform, calponin, desmin, smooth muscle myosin heavy chain, myosin light chain kinase, and β-tropomyosin decays. On the other hand, the content of non-muscle myosin heavy chain, low-molecular-weight caldesmonlMW, vimentin, protein kinase-Cα and -Cβ, and CD44 heightens (1–6 fold) when cells divide [719]. The content in cytoskeletal proteins is greater in the trachealis muscle than that in the media of the pulmonary artery [719]. Airway smooth myocytes shorten faster than arterial ones.
8.4.3 Calcium Influx
Plasma membrane depolarization activatesCaV channels. Subsequent Ca2 + entry causes rapidly (in a few milliseconds) local Ca2 + release through ryanodine receptors from Ca2 + store in myocytes.22 All 3 types of ryanodine receptors (RyR1–RyR3) are expressed in smooth myocytes.
In airway smooth myocytes, membrane depolarization does not prime instantaneous Ca2 + release. However, voltage depolarization during sufficiently long duration ( > 1 s) can induce local Ca2 + release through ryanodine receptors, independently of CaV channels and associated extracellular Ca2 + import into the cytosol [720].
ResultingCa2 + sparks prime cell contraction. The long latency for depolarization-induced Ca2 + release in airway smooth myocytes is explained by involvement of GPCR that can serve as voltage sensors and their initiated signaling. This type of mechanism is illustrated by activatedM3 muscarinic receptors in the absence of exogenous agonists that trigger the Gq–PLC–IP3 axis. This signaling pathway provokes a Ca2 + release through IP3Rs from the sarcoplasmic reticulum that is then enhanced by that through RyR2 (local Ca2 + -induced Ca2 + release), but neither RyR1 nor RyR3 [720].
8.4.4 Airway Smooth Myocyte Products
Airway smooth myocytes synthesize growth factors, cytokines, and chemokines, as well as lipid mediators, in addition to receptors, ion carriers, enzymes, and constituents of the extracellular matrix (Table 8.5).
Table 8.5
Examples of molecules synthesized by airway smooth myocytes, such as growth factors, including cytokines and chemokines, as well as receptors and enzymes (Main source: [739]; A i : adenosine receptor; AR: adrenergic receptor; B2 bradykinin receptor; CCL: chemokine CC-motif ligand; COx: cyclooxygenase; CSF: colony-stimulating factor; CysLT: cysteinyl leukotriene receptor; ECM: extracellular matrix; ETR: endothelin receptor; H1: histamine receptor; ICAM: intercellular adhesion molecule; Ifnγ: interferon-γ; Ifnγ R: Ifnγ receptor; ILi: interleukin type-i; ILiR: ILi receptor; K Ca 1.1: voltage-sensitive, calcium-activated large conductance potassium channel (BK); LIF: leukemia inhibitory factor [a.k.a. cholinergic differentiation factor (CDF) and differentiation-stimulating factor (DIA)]; LPAR: lysophosphatidic acid receptor; mAChR: muscarinic acetylcholine receptor; MHC: major histocompatibility complex; NKR: neurokinin receptor; NOS: nitric oxide synthase; PAR: peptidase-activated receptor; PDGF: platelet-derived growth factor; PGe2: prostaglandin-E2; PGi2: prostacyclin; sPLA2: secreted phospholipase A2; PR: prostanoid receptor; RAR: retinoic acid receptor; RXR: retinoid X receptor; S1PR: sphingosine 1-phosphate receptor; SCF: stem cell factor; VCAM: vascular cell adhesion molecule; VIPR: vasoactive intestinal peptide receptor; 5HTR: serotonin receptors).
Type | Molecules |
|---|---|
Growth factors | IGF2, PDGFa, PDGFbb, SCF, VEGFa |
Cytokines | CSF2, IL2/5/6/8/11/12, Ifnγ, LIF |
Chemokines | CCL2/5/7/8/11 |
Receptors | A 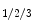 , AR, B2, CysLT, ETR, H1, , AR, B2, CysLT, ETR, H1, |
LPAR, mAChR, NKR, PAR, PR, S1PR, VIPR, 5HTR | |
EGFR, IGFR1/2, PDGFRα/β, | |
IL2R, IL12R, IL13R, Ifnγ R, TNFRSF5, | |
CCR3, Class-2 MHC, CD44 | |
Adhesion molecules | ICAM1, VCAM1 |
Lipid mediators | PGe2, PGi2 |
Enzymes | COx1/2, MMP1/2, NOS, sPLA2 |
Ion carriers | IP3R, RyR, SERCA, CaV1/3 |
K IR 2.1, K Ca 1.1 | |
ECM components | Tenascin-C (during healing) |
Nuclear receptors | RARα/β/γ, RXRα/β |
Airway remodeling relies on changes in phenotype of airway smooth myocytes and fibroblasts, especially inasthma. Mitogens are released by epithelial cells, fibroblasts, smooth myocytes, mastocytes, and eosinophils. In addition to mechanical stress, several growth factors can be involved in the change in aSMC phenotype, such as epidermal (EGF), fibroblast (FGF2), and platelet-derived (PDGF) growth factors.
8.4.4.1 G-Protein-Coupled Receptors
In humans, airway smooth myocytes are endowed with multiple types ( > 350 [721]) ofG-protein-coupled receptors that participate in the intra-, auto-, juxta-, and paracrine regulation. Furthermore, alternative splicing diversifies the recepterome and augments the number of GPCR types encountered on their surface, hence heterogeneity in GPCR signaling. More than 190 GPCRs have, on average, 5 different receptor isoforms that result from various types of alternative splicing of pre-mRNAs, such as alternative splice donors and acceptors, novel introns, intron retentions, exon(s) skips, and novel exons [721].
In airways,GPCRs expressed on smooth myocytes contribute to the regulation of contraction and relaxation state. Histamine, leukotriene, prostaglandin, and muscarinic Gq-coupled receptors support contraction, whereas Gs-coupled β2-adrenergic receptors foster relaxation.
In addition, in airway smooth myocytes, G-protein-coupled receptors modulate secretion of growth factors and other signaling mediators such as eicosanoids that intervene in airway inflammation (Table 8.6).
Table 8.6
G-protein-coupled receptors and their effects in airway smooth myocytes (Source: [722]; A i : adenosine receptor; AR: adrenergic receptor; B i : bradykinin receptor; CysLT i : cysteinyl leukotriene receptor; EP i : prostanoid receptor; ET x : endothelin receptor; H i : histamine receptor; IP: prostanoid (prostacyclin) receptor; LPA i : lysophosphatidic acid receptor; M i : muscarinic acetylcholine receptor; NK i : neurokinin receptor; PAR i : peptidase-activated receptor; RasGRF: Ras guanosine nucleotide-releasing factor [RasGEF]; RasGRP: Ras guanosine nucleotide-releasing protein [RasGEF]; S1P i : sphingosine 1-phosphate receptor; TP: prostanoid (thromboxane A2) receptor; VIPR: vasoactive intestinal peptide receptor; 5HT i : metabotropic serotonin (or 5-hydroxytryptamine) receptors).
GPCR | Main G protein | Function |
|---|---|---|
Type | partner | |
5HT2C | Gq | PLC–IP3–Ca–RasGRF1–Ras–ERK1/2 axes |
Gi | Contraction, growth potentiation | |
α1AR | Gq | |
β2AR | Gs | Relaxation, growth inhibition, |
regulation of cytokine synthesis | ||
A1 | Gi | Contraction |
A2B | Gs | Relaxation |
A3 | Gq | |
B2 | Gq | Contraction |
CysLT1 | Gq | Contraction |
EP2 | Gs | Relaxation, growth inhibition, |
regulation of cytokine synthesis | ||
IP | Gs | Growth inhibition |
TP | Gq | Contraction, growth potentiation |
ETA–ETB | Gq | Contraction, growth potentiation |
H1 | Gq | Contraction, growth potentiation |
LPA1–LPA4 | Gq, Gi, G12/13 | Growth stimulation, |
S1P1–S1P3 | Gq, Gi, G12/13 | Regulation of cytokine synthesis |
M2 | Gi | |
M3 | Gq | Contraction, growth potentiation |
NK1–NK2 | Gq | Contraction, growth potentiation |
PAR1–PAR3 | Gq, Gi, G12/13 | Growth stimulation and potentiation |
P2Y | Gq | |
VIPR | Gs | Relaxation, growth inhibition |
Rapid desensitization results from receptor phosphorylation byG-protein- coupled receptor kinases (GRK). Arrestin binding mediated by GRKs initiates receptorendocytosis for resensitization after recycling to the plasma membrane. In addition, activated intracellular kinases, such as protein kinases PKA and PKC also phosphorylate GPCRs and lead to GPCR–G-protein decoupling.
RasGRF Activators
The RasGRF proteins are coupled to G-protein-coupled receptors. Three main families of guanosine nucleotide-exchange factors (GEF) are associated withRas GTPases and link plasmalemmal receptors to Ras protein activation:Son of sevenless (SOS1–SOS2),Ras guanosine nucleotide releasing factors (RasGRF1–RasGRF2), andRas guanosine nucleotide releasing proteins (RasGRP1–RasGRP4; Vol. 4 – Chap. 9. Guanosine Triphosphatases and Their Regulators). Activators of the SOS and RasGRP sets link (via GRB2 adaptor andphospholipase-Cγ-dependent generation of diacylglycerol, respectively) membrane or cytoplasmic protein Tyr kinases to Ras GTPases. Calcium influx promotes RasGRF translocation to the plasma membrane, where it can be phosphorylated (activated).
Gq-Coupled Receptors and Bronchoconstriction
Other Gq-coupled receptors that can cause contraction include A3 adenosine, B2 bradykinin, ETA endothelin, H1 histamine, NK1 and NK2 neurokinin, andP2 purinergic receptors [722]. Both thrombin and lysophosphatidic acid that operate via Gq-coupled receptors PARs and LPARs provoke cell proliferation. Endothelin, histamine, leukotriene-D4, sphingosine 1-phosphate, and thromboxane potentiate mitogenic effects of receptor protein Tyr kinases via their cognate Gq-coupled receptors.
High-Affinity Receptors for Leukotriene-B4
For example, high-affinity receptors for leukotriene-B4 (LTb4R), i.e., BLT1 and BLT2 (Vol. 3 – Chap. 7. G-Protein-Coupled Receptors), are expressed on neutrophils, eosinophils, and T lymphocytes. They promote leukocyte migration to the lung and participate in their activation and degranulation. Moreover, they are produced by human airway smooth myocytes. They are coupled to Gαi or Gαq subunit. Three forms of the transcribed LTb4R and 2 additional variations in the 5′ UTR exist [721].
Gs-Coupled Receptors and Bronchodilation
Gs-coupled receptors elicit relaxation of airway smooth myocytes. They compriseβ2 adrenergic,A2B adenosine,prostanoid EP2 and IP (for prostaglandin-E2 and prostacyclin, respectively), and vasoactive intestinal peptide(VIP) receptors [722]. Activated Gs stimulates membrane-boundadenylate cyclase AC5 and AC6 isoforms. Activated adenylate cyclases create second messenger cAMP from ATP to activateprotein kinase-A. This enzyme can phosphorylate some Gq-coupled receptors, phospholipase-C, as well as IP3Rs to prevent phosphoinositide generation and calcium influx. It also phosphorylatesmyosin light chain kinase, thereby reducing its affinity to calcium–calmodulin and activity. It phosphorylates calcium-activated potassium channel to cause K + efflux and hyperpolarization. It phosphorylates the transcription factorcAMP-responsive element-binding protein (CREB) to control the synthesis of numerous immunomodulatory proteins.
VIP Receptor
Vasoactive intestinal peptide (VIP), a relaxant neuropeptide, impedes translocation of cRaf to the plasma membrane induced by ceramide as well as that of PKCα [723]. The Ras-mediated signaling links plasmalemmal receptors tomitogen-activated protein kinase modules that phosphorylate (activate) several target proteins such as 27-kDaheat shock protein (HSP27). In smooth myocytes, HSP27 colocalizes with actin filaments and proteins of the contractile cytoskeleton, such as myosin, tropomyosin, and caldesmon to modulate the interaction between these proteins. Phosphorylation of HSP27 promotes actin polymerization and stress fiber formation. In airway smooth myocytes, phosphorylated HSP27 stabilizes the actin cytoskeleton [724].
Gi-Coupled Receptors
A third group of GPCRs preferentially couples to members of the Gαi family (Gi1–Gi3). Activated Gi hampers the activity of adenylate cyclases. On the other hand, Gi and/or Gβγ subunits stimulate Rho guanine nucleotide-exchange factors, hence Rho GTPases for actin polymerization. Dimer Gβγ inhibits AC1, but can enhance the activity of certain adenylate cyclase isoforms, such as AC2, AC4, and AC7, as well as sensitization of adenylate cyclases to stimuli [722]. In addition, Gβγ can activatephospholipase-Cβ.
G12/13-Coupled Receptors
A forth set of G-protein-coupled receptors associate with members of the G12/13 subclass. Their effectors encompass members of a family of guanine nucleotide-exchange factors for small Rho GTPases. G12/13-coupled receptors may includeLPA,S1P,peptidase-activated (PAR), andprostanoid TP (for thromboxane-A2) receptors [722].
Adrenergic Receptors
In human lungs, the amount of β-adrenoceptors is greater than that of α-adrenoceptors [725].
α-Adrenoceptors
Airway smooth myocytes synthesize both α1- and α2-adrenoceptors [725]. α-Adrenoceptors causebronchoconstriction (Table 8.7). α1a-Adrenoceptors primarily act on calcium channels, thereby fostering entry of extracellular Ca2 + ions. α1b-Adrenoceptors activatesphospholipase-C that processes phosphatidylinositol (4,5)-bisphosphate in the plasma membrane to form inositol (1,4,5)-trisphosphate and diacylglycerol, which triggers Ca2 + release from intracellular stores and activates Ca2 + – and phospholipid-dependent protein kinase-C. The latter may contribute to smooth muscle contraction by increasing the Ca2 + sensitivity of contractile proteins. α2-Adrenoceptors inhibit adenylate cyclase via guanine nucleotide inhibitory protein (Gi).
Table 8.7
α-Adrenoceptors in the lung and their function (Source: [725]). The tone of airway smooth myocytes is mainly regulated by members of the Gq/11 and Gs subclasses of G-protein subunits that cause bronchoconstriction and bronchodilation, respectively.
Site | Predominant | Effect |
|---|---|---|
subtype | ||
Airway smooth myocyte | α1 | Contraction |
Vascular smooth myocyte | α1 | Contraction |
Autonomic nerves | α2 | Neurotransmitter release decrease |
Secretory glands | α1 | Secretion increase |
Mastocyte | Mediator release increase |
Both noradrenaline and adrenaline increase very weakly the bronchial tone after pretreatment with β-adrenoceptor antagonists in bronchi isolated from healthy human lungs; the response is not enhanced in the presence of histamine [725].
β-Adrenoceptors
Both β1- and β2-adrenoceptors lodge on airway smooth myocytes ( ∼ 20 and 80%, respectively) in human lungs [725]; hence, in human bronchial smooth myocytes, β-adrenoceptors belong mainly to the β2-subtype. β-Adrenoceptors can also be detected in respiratory epithelial as well as mucous and serous gland cells of the trachea; however, most of the β-adrenoceptor pool of the human lung localizes in alveolar cells.
Gs-coupled β2-adrenergic receptors cause abronchorelaxation and inhibit proliferation of human airway smooth myocytes. β2-Adrenoceptors also mediate vasodilation and influence mucus production and transport and activity of cholinergic nerves [725].
Relaxation of airway smooth muscles results from Gs stimulation and subsequent elevation of intracellular cAMP level and activation of protein kinase-A. The latter phosphorylates myosin light chain kinase, thereby reducing MLCK affinity for calcium–calmodulin, myosin phosphorylation, and actin–myosin coupling. Smooth myocyte relaxation can also result from the decrease in concentration of free calcium ions. In fact, β2-adrenoceptors operate via cAMP andRapGEF3 and RapGEF4,23 but not protein kinase-A [726].
Muscarinic Acetylcholine Receptors
Airway smooth myocytes are innervated by postganglionic parasympathetic nerves that release acetylcholine. Muscarinic acetylcholine M2 and M3 receptors (mAChR) are coexpressed in many types of smooth myocytes, such as those in walls of blood vessels and airways.
The density of M2 receptors is greater than that of M3 receptors (relative density ≥ 4:1) [727]. In smooth myocytes, M2 receptor inhibits AC5 and AC6 adenylate cyclases via Gαi3, whereas Gq-coupled M3 activates AC5 and AC6 via Gβγ [728].
In airway smooth myocytes, activated Gi-coupled M2 receptor inhibits β-adrenergic-mediated relaxation; activated Gq-coupled M3 receptor initiates their contraction. Gq-coupled receptors are the main mediators of contraction of airway smooth myocytes. Neurotransmitter acetylcholine acts on M3 receptors to control the resting tone.
8.4.4.2 Receptor Protein Tyrosine Kinases
Epidermal Growth Factor Family Members
Bronchial epithelial cells produce members of the epidermal growth factor family, such as EGF, transforming TGFα and heparin-binding EGF-like (HBEGF) growth factors, and amphiregulin, that can stimulate cell migration, proliferation, differentiation, and survival, as well as repair of bronchial epithelia.
Bothepidermal (EGF) andheparin-binding EGF-like (HBEGF) growth factor promote repair of human bronchial epithelial cells [729].
In normal subjects, the concentration of epidermal growth factor receptor (HER1 or EGFR) is lower than that in asthmatic subjects [729]. The more severe the asthma, the higher the EGFR level. Agent EGF contributes to hypertrophy and hyperplasia of airway smooth myocytes during airway remodeling in asthma [730].
Insulin-like Growth Factor and Partners
Insulin-like growth factor IGF2, IGF-binding protein IGFBP2 (but neither IGF1 nor IGFBP1), and both IGF receptorsIGFR1 andIGFR2 are produced in airway smooth myocytes [732]. In addition, IGF2 and IGFBP2 are secreted.
Platelet-Derived Growth Factors
Platelet-derived growth factors (PDGF) are mitogens and recruitment factors for smooth myocytes. Protomer PDGFb signals via the receptor protein Tyr kinase PDGFRβ.24 Protomers PDGFa and PDGFb are synthesized by various cell types, including vascular smooth myocytes. Isotype PDGFa is required during the body’s development in the recruitment of smooth myocytes to alveolar sacs and their compartmentation into alveoli [733]. Smooth myocytes express both PDGFR types, but usually higher levels of PDGFRβ.
Vascular Endothelial Growth Factors
In humans, airway smooth myocyte synthesizesvascular endothelial growth factor VEGFa isoforms, such as VEGFa121, VEGFa165, VEGFa189, and VEGFa206 splice variants [731]. The constitutive secretion of VEGFa increases onbradykinin exposure, activation of B2 bradykinin receptor andprotein kinase-C, and generation of prostanoids. Activated cyclooxygenase-2 produces predominantlyprostaglandins PGe2 andprostacyclin (PGi2).25
8.4.4.3 Epican
Plasmalemmal CD44 glycoprotein, or epican, a heparan sulfate proteoglycan, is also synthesized by airway smooth myocytes. This molecule interacts with hyaluronic acid,collagens, andmatrix metallopeptidases; its glycosylated form with fibrinogen and fibrin; its sialyfucosylated glycoforms with selectins. Matrix metallopeptidase MMP2 is an autocrine factor required for airway smooth myocyte proliferation.
8.4.5 Hyperresponsiveness and Hypersensitivity
Asthma is characterized byhyperplasia andhypertrophy of airway smooth myocytes that strongly (hyperresponsiveness) contract in response to low-magnitude stimuli (hypersensitivity), in addition to tissue inflammation with edema and thickening of the subepithelial basement membrane. Both wall edema andbronchoconstriction narrow lumens of ventilatory ducts, thereby reducing air flow as demonstrated by lung function tests. Smooth myocytes colocalize withmastocytes. In addition, released interleukin-8 is a chemoattractant for neutrophils and eosinophils. Mastocytes do not affect the rate of proliferation, but prevent CCL11-mediated CCR3-primed migration of airway smooth myocytes [736].
Basement membrane thickening is due to the deposition of interstitial collagen-1, -3, and -5 as well as fibronectin. Collagens are synthesized bymyofibroblasts, the number and activity of which rise in asthma due to fibroblast (FGF2), insulin-like (IGF1), dimeric platelet-derived (PDGFbb), and transforming (TGFβ) growth factor, as well as endothelin-1 [737, 738].
Airway smooth myocytes contribute to the pathogenesis of asthma because of [739]: (1) proliferation beyond the ordinary rate provoked by mitogens and (2) recruitment of progenitors to airway smooth myocyte bundles, thereby causing hyperplasia in addition to hypertrophy; (3) interactions of airway SMCs with inflammatory cells, such as T lymphocytes and eosinophils; and (4) their secretion of pro-inflammatory cytokines, in particular chemokines (Table 8.5).26
Sphingosine 1-phosphate operates as an inflammatory mediator in asthma. It provokes airway smooth muscle contraction and growth as well as production of pro-inflammatory cytokines such as interleukin-6 to trigger bronchoconstriction and airway inflammation and remodeling [740].
8.4.6 Deep Inspiration as Bronchodilation Inducer
Deep inspiration stretches airway smooth myocytes, thereby softening, or fluidizing the cytoskeleton, especially stress fibers. Rapid fluidization followed by a slow resolidification enables cells to reorganize theirstress fibers and adhesion plaques in response to mechanical stresses [741]. Resulting heightened mobility of cytoskeleton constituents and partners generates a significantbronchodilation.
In asthma, this phenomenon disappears due to remodeling of the cytoskeleton and surrounding matrix [741]. However, in an acute, mild asthma attack, bronchospasm can be counteracted by a slow, complete inspiration. Deep inspirations that are too fast or delayed remain ineffective. In severe asthmatic crises, slow and complete inspirations do not relieve bronchospasm. The failure of deep inspiration-generated bronchodilation may cause airway hyperresponsiveness.
8.4.6.1 Contractile Cytoskeleton Adaptivity
Mural smooth myocytes have a large functioning length range, in opposition to striated myocytes that works at most at 20% of their resting length. The malleability of the myofilament lattice of smooth myocytes results from remodeling of contractile filaments of the cytoskeleton in response to stress and strain exerted on these cells [742].
Smooth muscle length is determined not only by the balance between active force that it generates and passive reaction developed by the external load, but also, due to the breathing time scale, by an outside-equilibrium behavior [743]. In normal conditions, tidal fluctuations of muscle length suffice to avoid bronchoconstriction.
The malleable cytoskeleton behavior is similar to that of soft, colloidal materials [743].27 The elastic modulus ranges from the order of 1 Pa to 1 kPa. The stiffness over a wide range of frequencies does not exhibit any characteristic molecular relaxation time or resonant frequency. Frictional stress within the network is typically smaller than elastic stress. Both stress types are scale-free, as material rheological responses are not associated with any particular relaxation time and rise with frequency. On application of shear of sufficient magnitude, these materials become fluid-like bodies. When shear is stopped, they return to their original solid-like state. Colloidal dispersions can actually exhibit both fluid-like and solid-like behavior, with various types of transition between these two states. Colloidal particles with an attractive interparticle energy display a fluid-to-solid transformation due to gelation, at a critical volume fraction that depends on the magnitude of attractive interparticle energy on contact. Gelation occurs when interconnected colloid-rich regions solidify. Other features of soft, glassy materials include the rheological pattern that results from the competition between the spontaneous restructuration (slow scale-free recovery of rheological properties, the so-called aging) and destruction of the internal structure by shear (“shear rejuvenation”). Intermittent motions result from nanoscale rearrangements. Sub- and superdiffusive displacements rely on interactions between material constituents. Material transfer creates to low- and high-density regions of given types of constituents. Slowing of dynamics and more prominent intermittency of structural rearrangements indicate approach of a transition and kinetic arrest of colloidal particles, when crowded particles are trapped by their nearest neighbors (gelation).
Smooth myocytes adapt to a wide range of cell lengths and maintain optimal contractility. Assembly and disassembly of cytoskeletal filaments in smooth myocytes enable adaptation to large changes in cell geometry. The ability of actomyosin filaments to form and dissolve within the myofilament lattice at adequate sites and suitable instants yields the basic mechanism of cell adaptation [742]. Formation of myosin filaments is promoted by various filament-stabilizing proteins such as caldesmon. Many proteins may be assigned, removed, or reassigned in the context of reversible reorganization of the cytoskeleton. Myosin filament length is determined by the size of the actin filament network [742].
In airway smooth myocytes, the amount of contractile filaments can change rapidly to accommodate the context. Actin filament content actually increases during contractile activation. Myosin filament content also varies. In relaxed smooth myocytes, the density of myosin thick filaments is low, although of significant number. Myosin filaments indeed undergo rapid cycles of assembly and disassembly. Upon contractile stimulation, the content of myosin filaments in airway smooth myocytes rises strongly [742]. In solution, assembly of non-phosphorylated myosin monomers into filaments depends on monomer concentration. Once a critical concentration is reached, self-assembly occurs. The critical concentration increases when ATP is available and decreases when the myosin regulatory light chain is phosphorylated. In addition, C-terminal isoforms SM1 and SM2 of smooth muscle myosin heavy chain influence stability of filament assembly. Isoform SM1 confers a greater stability [742]. Yet, myosin filament lability is required for adaptive cell remodeling. Synthesis of SM1 and SM2 isoforms differs among different types of smooth myocytes as well as during development. Isoform SM2 may enhance SMC adaptation to large changes in length. In the presence of ATPMg, phosphorylation of the myosin regulatory light chain as well as other facilitating factors in smooth myocytes may regulate thick filament assembly.
Upon large-amplitude length oscillation applied to relaxed trachealis muscle strips, myosin filaments can partially disassemble [742]. These filaments reassemble when smooth muscle is repeatedly activated isometrically, following phosphorylation of regulatory myosin light chains. Moreover, myosin filament formation increases in the presence of actin filaments. Caldesmon that is detected interspersed in the actin filament network where myosin filaments are located can also provoke reassembly of long myosin filaments.
8.4.6.2 Cell Mechanoresponsiveness
Every cell exerts traction on its matrix and experiences more or less large time-dependent stretch exerted by its environment. The cytoskeleton can stiffen, hence increasing traction (reinforcement response), or can soften (fluidization response) [744]. Therefore, cells respond to imposed stretch using 2 procedures. (1) Reinforcement refers to rapid actin polymerization and increased focal adhesion assembly that cause heightened cytoskeletal stiffness and traction forces. Actin polymerization and action of nanomotors can indeed generate a stress field and traction forces. However, in cells that bear cyclically significant stretch (e.g., cells of heart and lung), cell reinforcement that causes cell stiffening thus progressively impedes normal organ deformation during the functioning cycle. Hence, the reinforcement response corresponds to an inadequate strategy. These cell types then select the opposing compensatory mechanism. Fluidization means prompt reduction in cytoskeleton stiffness and elevation in molecular mobility.
When they are subjected to repetitive homogeneous stretch, human airway smooth myocytes rapidly soften, fluidize, and decrease traction force, with subsequent slow recoveries [744]. After completion of a transient stretch–unstretch maneuver of 4-s duration, the traction field quickly decays and slowly recovers. In addition, in substrates of different stiffness, the greater the matrix stiffness, the larger the static traction. On the other hand, cell response to dynamic traction is similar, whatever the matrix stiffness [744]. Therefore, dynamic responses to stretch are governed by a bulk mechanism with a time scale of the order of 1 s that does not consider changes in substrate stiffness. In addition, uni- and biaxial, isotropic stretches cause identical fluidization responses. Whereas homogeneous stretch induces fluidization, heterogeneous stretch induces reinforcement. Although fluidization response does not depend on stretch homogeneity. subsequent recoveries markedly differ [744]. Cell response is influenced by the duration of transient stretch. Traction recovery after a homogeneous biaxial stretch of 4-s duration never exceeds the prestretch value, whereas traction recovery following a homogeneous stretch of 30-s duration overtakes it. However, this reinforcement response disappears when homogeneous biaxial transient stretches are applied repetitively. On the other hand, repetitive, heterogeneous, transient stretches generate upon load removal a rapid fluidization followed by reinforcement [744]. Other smooth myocyte types as well as human umbilical vein endothelial cells exhibit the same behavior.
In summary, in repetitive homogeneous cell stretch, whether isotropic or not, the fluidization response prevails. The reinforcement response to repetitive load transients in human airway smooth myocytes occurs in the case of heterogeneous cell deformations. Transient stretch causes [744]: (1) a quick detachment of the myosin nanomotor from actin; (2) a transient decline in Factin concentration; and (3) a change in interactions among cytoskeletal proteins.
8.5 Vascular Smooth Myocytes
Vascular smooth myocytes are implicated in the maintenance of vessel wall, in the regulation of blood pressure and flow, and response to wall damage. Mature smooth myocytes are not terminally differentiated because they can change their phenotype.
Vascular smooth myocytes are responsible for vasomotor tone using actin–myosin filaments. The contractile apparatus of differentiated smooth myocytes encompasses smooth muscle isoforms of α-actin, myosin heavy chain,calponin-1,28 transgelin-1, and smoothelin.
The vasomotor tone depends on sympathetic innervation and neurohumoral vasoactive command as well as various types ofmechanotransduction processes according to the region within the arterial compartment (large arteries to minimize cardiac postload, small arteries and arterioles to maintain a constant nutritive blood flow, and small arterioles to fit the local metabolism needs).
Arterial smooth myocytes regulate blood pressure and flow according to cardiac postload and tissue metabolism needs. In the microvasculature, smooth muscles construct precapillary sphincters that regulate blood flow in capillary beds.
Vascular smooth myocytes include phasic and tonic varieties. The tonic type does not generate action potentials, whereas the phasic type in the portal system and lymphatics creates contraction waves to propel luminal fluid toward the heart.
Vascular smooth myocytes also differ according to vascular compartment (whether they pertain to an artery or a vein, to a large artery or resistance arteriole), embryological origin, and organ-dependent microenvironment.
Due to blood pressure-dependent radial hydraulic conductance, the arterial circulating plasma molecules are outwardly convected. Transferred macromolecules within the vessel wall act as extrinsic stimuli of mural cells. Vascular smooth myocytes can reprogram their expression pattern in response to acute and chronic stimuli. In addition, mechanical signals can directly (blood pressure-derived intramural tension) or indirectly (shear rate-induced endothelial command) influence the vSMC phenotype. In particular, arterial smooth myocytes respond to hypertension and resulting increase in wall tension by hypertrophy and secreting extracellular matrix constituents via RhoA, ERK1, and ERK2 [745].
Differentiated vascular smooth myocytes are also able to modify their phenotype in response to vessel wall inflammation or injury. In healthy vasculature, smooth myocytes remain quiescent and highly contractile; in disorders of the arterial wall, smooth myocytes adopt a proliferative phenotype and loose their non-contractile phenotype. Vascular diseases (aneurysm, atherosclerosis, hypertension, and poststenting restenosis; Vol. 6 – Chap. 8. Vascular Diseases) are generally characterized by smooth myocyte phenotype switching. In response to vessel damage, smooth myocytes dedifferentiate and causeintimal hyperplasia. They proliferate, migrate from the media to intima, secrete remodeling enzymes such as metallopeptidases, and increase matrix protein synthesis, but decrease the production of contractile proteins (smooth muscle α-actin, smooth muscle myosin heavy chain, transgelin).29
8.5.1 Vascular Smooth Myocyte Subtypes
Several subpopulations of smooth myocytes (length 20–60 μ m, center width ∼ 4 μ m) can be described in the arteries, such as spindle-shaped smooth myocytes (sSMC) and rhomboid SMCs (rSMC). The SMC morphology can be modulated by growth factors and environmental stimuli [746]. A large proportion of rSMCs is observed after stent implantation (Vol. 6 – Chap. 10. Treatments of Cardiovascular Diseases), with high proliferative and migratory activities.
8.5.2 Mesangial and Juxtaglomerular Cells
In kidneys,mesangial cells are smooth muscle-like cells that participate in the formation of glomeruli of nephron entries. The glomerulus filters blood supplied by an afferent arteriole that drains into an efferent arteriole to yield urine into the Bowman’s capsule, the basic filtration unit of nephron. The blood–urine filtration barrier is constituted by endothelial cells of the glomerulus, negatively charged glomerular basement membrane (thickness 250–350 nm), and podocytes of the Bowman’s capsule that control the filtration of proteins. Glomerular mesangial cells lodge in the interstitium between endothelial cells. They control the filtration surface area and the local blood flow, as they can contract in response mainly to stretch. They also secrete extracellular matrix constituents, prostaglandins, and cytokines. They scavenge molecules entrapped in the glomerular basement membrane.
Specialized mural, granular smooth myocytes, the so-called juxtaglomerular cells, reside in the wall of the afferent arteriole of each nephron. The juxtaglomerular apparatus localizes between the vascular pole of the renal corpuscle (initial blood-filtering component of the nephron with the glomerulus and Bowman’s capsule) and the distal convoluted tubule of the nephron. Juxtaglomerular cells synthesize, store, and secreterenin. Renin activates the angiotensin cascade to regulate blood volume and pressure. Juxtaglomerular cells are endowed withβ1-adrenergic receptors. Once stimulated by cathecholamines, these receptors provoke renin secretion.
8.5.3 Vascular Smooth Myocyte Phenotypes
Vascular smooth myocytes have various subtypes that prevent easy identification in histological sections. Moreover, their fate oscillates from quiescent, differentiated, contractile cells to proliferative cells, according to types of local environmental signals (Table 8.8). The vSMC phenotype is controlled by interactions between general and SMC-specific factors. Phenotype change is a rapid adaptation to fluctuating environmental cues.
Table 8.8
Vascular smooth myocyte phenotypes (Sources: [747, 748]). These cells can reprogram their expression pattern according to environmental conditions. These changes can trigger chromatin remodeling especially using acetylation and methylation of histones that modifies DNA accessibility to transcription factors, such as Krüppel-like factor KLF4, P300, myocardin, and serum responsive factor (SRF). In fact, numerous chromatin regulators act at SMC-specific marker sites to silence or enable access to the transcriptional machinery. Various stimuli influence the SMC phenotype, such as transforming growth factor-β, platelet-derived growth factor, retinoic acid, and oxidized phospholipids.
Phenotype | Features |
|---|---|
Contractile | Synthesis of contractile proteins |
quiescent | Low proliferative rate |
Synthetic | Production and secretion of matrix constituents |
(matrix) and | and metallopeptidases |
proliferative | Interplay between KLF4, KLF5, and miR146a |
Endocytic | Uptake of modified LDLs in atheroma |
and | Pinocytosis of small components of the fluid phase |
phagocytic | Phagocytosis of molecular complexes, cell particles, |
or apoptotic cells | |
Organizer of | Acquisition of ability to migrate and proliferate |
advential | Production of inflammatory signals |
response | |
Osteoblastic | Calcification in atheroma |
Vascular smooth myocyte reprogramming relies onchromatin remodeling that results from acetylation and methylation ofhistones.30
Expression of vSMC-specific genes is regulated by interactions of multiple transcription factors that are either ubiquitous (e.g., serum response factor) or specific of smooth myocytes (e.g., myocardin). The activity of these transcription factors and cofactors is regulated by kinases (CamK, ERK, JNK, P38MAPK, RoCK,31 and PKB) [745].
Factor KLF4 represses SMC marker genes. Phosphorylation of KLF4 and its subsequent acetylation byP300 enable SMC differentiation. Proliferative stimuli cause KLF4 dephosphorylation and deacetylation byHDAC2, association withELk1, ejection ofSRF, and repression of SMC markers.
Myocardin acts as a transcriptional SRF coactivator. Histone acetyltransferase P300 is involved not only in skeletal and cardiac myogenesis, but also in SMC differentiation. Gene activation by myocardin-SRF is enhanced upon P300 binding. Enzyme p300 can also enhance myocardin activity independently of SRF factor. Myocardin-related transcription factorsMRTFa and MRTFb are needed in SMC differentiation.
In fact, numerous chromatin regulators act at SMC-specific marker sites to silence or enable access to the transcriptional machinery. Various stimuli influence the SMC phenotype, such as transforming growth factor-β, platelet-derived growth factor, retinoic acid, and oxidized phospholipids.
Retinoic acid that fosters SMC differentiation targetsRARα andRXRα receptors; protein arginine (R) methyltransferase PRMT2 bind to these receptors and interacts nuclear receptor coactivator NCoA1,32 another histone acetyltransferase. On the other hand, PDGFbb and oxidized phospholipids are dedifferentiating stimuli that causes histone deacetylation and demethylation in SMC marker CArG-box chromatin.
In the differentiated state, they express high concentrations of smooth muscle-specific isoforms of contractile proteins. In the proliferative state, they secrete large amounts of extracellular matrix components and synthesize low levels of characteristic isoforms of contractile proteins. The biomarker smooth muscle α-actin does not necessarily identify a smooth myocyte because it is also produced by myofibroblasts.
Vascular smooth myocyte reprogramming results from rapid adaptation associated with protein turnover. The ubiquitin–proteasome system participates in rapid turnover of short- and medium-lived proteins via degradation of mediators involved in cell signaling,33 especially during cell differentiation and proliferation, that become unnecessary. Inhibition of cell cycle-controlling ubiquitin ligases or proteasome represses vSMC proliferation and prevents change to the synthetic phenotype [749].
When theendocytic phenotype is achieved, cationic proteins are mainly endocytosed by an absorptive mechanism, involving interactions with the negative charges of the glycocalyx; anionic proteins such as LDLs interact mainly with LDLR receptors and modified LDLs with scavenger receptors.
In atherosclerosis characterized by an osteoblastic phenotype, foci of calcification are likely linked to cell death and membrane–annexin-rich particle release, which supports calcium–phosphate concentration; extracellular free DNA triggers precipitation.
8.5.3.1 Mechanical Signals in Phenotype Change
Regulation of cell functions, such as proliferation and differentiation, relies not only on chemical compounds such as growth factors, but also on physical andmechanical signals, in particular geometrical and rheological features of the extracellular matrix and eventual anisotropy that influence cell spreading and curvature.34
Deformations of the plasma membrane activates mechanosensitive immersed proteins, such as mechanically gated ion channels, receptors, and cell adhesion molecules. In particular, integrins that cluster to build focal adhesions tether the intracellular actin cytoskeleton to the extracellular matrix. Focal adhesions are signalling hubs that contain kinases, such as FAK, MAPKs, and Src, and small GTPases such as Ras. The actin cytoskeleton also connects to the nucleus and its nucleoskeleton with intermediate filaments and lamins, via nestins (Vol. 1 – Chap. 4. Cell Structure and Function). Nuclear strains and stresses exerted on nuclear lamins and other nucleoskeletal constituents and changes in interactions of nuclear structural proteins with chromatin may modulate the chromatin organization and interplays with DNA-binding proteins, thus controlling gene expression patterns. Furthermore, mechanical stresses are converted into chemical signals that can be transmitted down to to the nucleus to modify gene expression.
In particular, effectors of theHippo pathway, or kinase (STK3 and STK4) cassette, Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ; or WWTR1) relay mechanical cues to the nucleus [750]. The regulation of YAP and TAZ mediators requiresRhoA GTPase and actin–myosin contractility. Transmitter YAP precludes expression of the contractile phenotype of smooth myocytes, as it impedes activity of the myocardin–SRF complex.
8.5.3.2 Differentiation of Vascular Smooth Myocytes
Chromatin remodeling enables SMC differentiation, in particular demethylation of dimethylated Lys9 of histone-3 (H3K9me2) and Lys20 of histone-4 (H4K20me2) that allows decompaction of SMC marker gene chromatin in SMC precursors [748]. Differentiated vascular smooth myocytes possess several specific histone tail modifications (acetylation [H3K9ac, H3K14ac, and H4ac) and methylation (H3K4me2, H3K79me2) at SMC marker gene CC(A/T-rich)6GG (CArG) boxes. These histone modifications enable access of themyocardin–SRF complex and coactivatorP300 histone acetyltransferase to SMC-specific CArGs, thereby activating gene transcription. Dedifferentiation of vascular smooth myocytes (phenotype change) results from removal of these histone modifications from chromatin at SMC marker gene CArG boxes, except H3K4me2 that may help maintain SMC marker genes as euchromatin and permit redifferentiation under adequate conditions [748].
During embryo- and fetogenesis, vascular smooth myocytes have various sources (e.g., neural crest, somites, and proepicardium, in particular mesoangioblasts of the dorsal aorta in mammals, in addition to endothelial cells upon endothelial–mesenchymal differentiation), according to the vascular compartment, in addition to a common population of progenitor cells, such as TIE1 + , PECAM1low, Cdh5 − , PTPRc − precursors [751]. These precursors also express smA and homeodomain-containing factor Msx2.
Notch and Wnt control vSMC differentiation and modulate their phenotype according to exerted stimuli via transcription factors and cofactors that determine gene expression program.
Notch signaling in TIE1 + precursor cells is involved in developing arteries, whatever the vascular compartment, but not in mature vascular smooth myocytes, i.e., once the artery is stabilized. Notch upregulates transcription of the mesenchymal markers smA and PDGFRβ; it supports endothelial–mesenchymal transdifferentiation.
8.5.3.3 Notch Signaling in vSMC Specification
Smooth myocytes of major arteries arise from several cell lineages. Smooth myocytes of the developing arch artery, ductus arteriosus, and proximal regions of the major aortic arch branches derive from the cardiac neural crest; those of the root of the aorta and pulmonary artery from the second heart field; those of the the descending aorta from somites [752].
Smooth myocytes of organ arteries have also diverse origins. The neural crest gives rise to smooth myocytes and pericytes of most brain vessels; the mesothelium and proepicardium generate mesenteric and coronary vessels, respectively [752].
Notch stimulates various aspects of vSMC specification, differentiation, and maturation during development. Notch positively regulates differentiation of cardiac neural crest precursor cells into smooth myocytes. Notch is also required in the second heart field for proper patterning of the outflow tract [752]. Notch controls the differentiation of epicardium-derived cells into coronary smooth myocytes.
In classical Notch signaling, the extracellular domain of Notch receptor on the surface of signal-receiving cells interacts with the extracellular domain of Notch ligand on apposed signal-sending cells (Vol. 3 – Chap. 10. Morphogen Receptors). Four Notch receptors (Notch-1–Notch-4) on signal-receiving smooth myocytes tether to 5 transmembrane Delta-like and Jagged ligands (DLL1, DLL3–DLL4, and Jag1–Jag2) on apposed signal-sending endothelial and smooth muscle cells. Notch-1 and Notch-4 reside primarily in endothelial cells; Notch-1 pulmonary artery smooth myocytes during development; Notch-2 in mesenchymal cells and aorta and pulmonary artery smooth myocytes; Notch-3 in smooth myocytes and pericytes of the microvasculature of the central nervous system [752]. Ligands DLL1, DLL4, and Jag2 lodge on endothelial cells; Jag1 on both endothelial and smooth muscle cells. In addition, Notch receptors can interact with other ligand types.35
Notch receptor is transported to the cell surface as a heterodimer. Notch receptor precursor is actually cleaved by furin during transport to the plasma membrane into a large ectodomain (NotchECD) and a membrane-tethered intracellular domain (NotchMTIC) that remain connected by a juxtamembrane heterodimerization domain.
Upon ligand binding, Notch is successively cleaved byadamlysins such as ADAM17 at the extracellular juxtamembrane region and by the γ-secretase complex that liberates the Notch intracellular domain (NotchICD) from the plasma membrane. On the one hand, NotchECD isendocytosed into the signal-sending cell. On the other, NotchICD translocates to the nucleus, where it forms an active transcriptional complex with RBPJκ transcription factor and coactivators such as Mastermind-like coactivator (MamL). In the absence of Notch, RBPJκ represses transcription, as it recruits histone deacetylases and other corepressors. Nuclear NotchICD displaces the histone deacetylase–corepressor complex from RBPJκ, enabling transcriptional activation of Notch target genes such as class-B basic helix–loop–helix proteins Hairy enhancer of Split (HES1–HES7 or bHLHb37–bHLHb43) and HES-related transcriptional regulators (HRT1–HRT3). In addition, other Notch target includes PDGFRβ, the microRNA cluster miR143–miR145, and components of the Notch pathway (e.g., Notch-3 and Jag1).
Intimal hyperplasia is characterized by a temporospatial change in Notch-1 to Notch-3 activity [752]. In smooth myocytes, Notch-1, but not Notch-3, mediates proliferation, migration, and survival, hence neointima formation.
Pulmonary arterial hypertension results from a remodeling of small pulmonary arteries with proliferation of smooth muscle and endothelial cells that causes wall thickening and lumen narrowing. The Notch-3-HES5 pathway is involved [752].
Platelet-derived growth factor-BB and transforming growth factor-β mediate vSMC phenotype switching. The former is involved in the repression of vSMC-specific gene expression; the latter promotes the contractile phenotype. Notch cooperates with PDGF and TGFβ to regulate vSMC migration and differentiation [745]. On the other hand,Wnt inhibitory factor-1 hampers PDGFbb-induced vSMC proliferation.
8.5.3.4 Wnt Signaling in vSMC Specification
Secreted, cysteine-rich Wnt glycoproteins that are encoded by 19 genes regulate SMC proliferation, differentiation, migration, and survival. They interact with 10 Frizzled receptors as well as coreceptors, such as low-density lipoprotein-related proteins LRP5 and LRP6 for the canonical, β-catenin dependent Wnt pathway and receptor protein Tyr kinase-like orphan receptor ROR2 for the non-canonical, β-catenin independent Wnt axis.
Wnt proteins regulate the synthesis of several matrix constituents, such as fibronectin and versican, as well as matrix metallopeptidases (MMP2, MMP3, MMP7, MMP9, MMP13, MMP14, and MMP26).
In the conventional pathway, β-catenin translocates to the nucleus, where it assists in the TCF–LEF-mediated transcription of target genes. In arterial and venous smooth myocytes, the β Ctn–TCF axis upregulates cyclin-D1 and downregulates CKI1a cyclin-dependent kinase inhibitor, thus increasing the proliferative rate. Wnt1 and Wnt3a can trigger cyclin-D1 production in arterial smooth myocytes in vitro. Under growth factor stimulation, Wnt4 supports arterial SMC proliferation in vitro; it also contributes to pathological intimal thickening in vivo [753].
In unconventional pathways, Wnts initiate the PLC–Ca2 + axis, thereby activating via calmodulin CamK2 and PP3 that stimulate NFκ B and NFAT transcription factors on the one hand and PKC that stimulates NFκ B and CREB transcription factors on the other. Furthermore, Wnt can bind to ROR2 in the absence of Frizzled to release Ca2 + from intracellular stores and activate JNK kinase. In addition, Wnt can also activate Rho and Rac GTPases, hence RoCK on the one hand, and PKA, thus CREB on the other. Factor NFAT1 promotes cyclin-D1 gene expression, hence arterial SMC proliferation [753]. In addition, CamK2 favors arterial SMC proliferation. Moreover, JNK is activated in proliferating smooth myocytes.
The canonical Wnt pathway is controlled by Dickkopf inhibitors that preclude Wnt–LRP interaction. Both canonical and non-canonical Wnt pathways are inhibited by secreted Frizzled-related proteins and Wnt inhibitory factor-1 that prevent Wnt-Fz linkage. Nevertheless, Dickkopf-1 promotes SMC proliferation in rat mesenteric microvasculature [753].
Differentiation of venous and arterial smooth myocytes may also be regulated by Wnt proteins. Wnt3a promotes the expression of the early myofibroblast marker SM22α; Wnt7b may intervene during differentiation of pulmonary smooth myocytes [753].
8.5.3.5 Proliferation of Vascular Smooth Myocytes
MicroRNAs contribute to the regulation of vascular smooth myocyte fate. The microRNA cluster miR143–miR145 as well as miR21 and miR1 support the contractile phenotype. On the other hand, miR221, miR146a, miR26a, and miR24 promote cell proliferation after vascular injury [745]. The microRNA cluster miR143–miR145 may be activated by Jagged-1–Notch andmyocardin–SRF axes. In young hypertensive patients, the expression of angiotensin AT1receptor is negatively correlated with miR155 agent.
Krüpple-like factors KLF4 and KLF5,ETS-like transcription factor ELk1, HES-related repressor protein HERP1,FoxO4, and RelA subunit ofNFκ B hinder vSMC-specific gene expression. These mediators propagate effects ofPDGFbb,TGFβ, and Notch, at least partly by disrupting SRF–myocardin interaction, thereby supporting the synthetic and proliferative phenotype of vascular smooth myocytes (Table 8.9) [745]. MicroRNA-143 and -145 are targets of SRF and myocardin, which maintain the contractile phenotype of vascular smooth myocytes and target KLF4 and ELk1 transcription factors.
Table 8.9
Control of phenotype of vascular smooth myocytes (Source: [745]). The synthetic, proliferative phenotype can result from increases in angiotensin-2, intracellular Ca2 + level, or stretch. On the other hand, the serum response factor (SRF)–myocardin complex maintains the contractile state. Cell contractility is associated with Ca2 + ion and RhoA signaling. Signaling initiated by Wnt and MAPK determines cell shape and promotes cell differentiation. Elongation of smooth myocytes supports the contractile phenotype.
Controllers | Mediators |
|---|---|
PDGF | KLF4, ELk1 |
TGFβ | KLF4, ELk1 |
Notch | HERP1 |
Wnt | MAPK |
Stretch | RhoA |
Calcium channels | Calcium |
Angiotensin-2 | RhoA |
MicroRNA-146a targets the 3′-untranslated region of the Krüppel-like factor Klf4 transcript, thereby promoting vSMC proliferation in vitro andintimal hyperplasia in vivo [754].36 Conversely, Krüppel-like factor KLF4 binds to and impedes activity of the miR146a promoter (mutual inhibition withfeedback loop), in competition with its KLF5 antagonist that promotes vSMC proliferation via miR146a regulator.37
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


