1999
Discovery of family with SQTS (Bjerregaard P, 1999, personal communication)
2000
Paper describing SQTS (Gussak et al. [1])
2003
High incidence of SCD in families with SQTS (Gaita et al. [2])
ICD treatment of patients with SQTS (Schrimpf et al. [3])
2004
Mutation found in families with SQTS (Brugada et al. [4])
Pharmacological treatment of patients with SQTS (Gaita et al. [5])
Experimental model of SQTS (Extramiana et al. [6])
2005
Review article of mechanism, diagnosis and treatment of SQTS (Bjerregaard et al. [7])
Successful prevention of SCD by an ICD in patient with SQTS (Schimpf et al. [8])
SQTS presenting as bradycardia in utero (Hong et al. [9])
SQTS website: shortqtsyndrome.org (Bjerregaard and Collier [10])
2006
Publication of the prevalence of a very short QT interval in the general population (Gallagher et al. [11])
2007
Overlap syndromes of SQTS and Brugada Syndrome (Antzelevitch et al. [12])
Mutation linking SQTS to SIDS (Arnestad et al. [13])
Population study of prevalence and prognostic significance of a short QT interval (Anttonen et al. [14])
2008
Animal model: Zebrafish with SQTS (Hassel et al. [15])
2009
Safety issue warning regarding drug induced shortening of the QT/QTc interval (Holbrook et al. [16])
2011
Diagnostic criteria to facilitate clinical recognition of SQTS (Gollop et al. [17])
Long-term follow-up of patients with SQTS (Giustetto et al. [18])
Definition and Terminology
By its own definition, any clinical syndrome is a combination of signs and symptoms that occur together and characterize a particular abnormality. In this context, short QT syndrome (SQTS) is best defined as an inheritable, primary electrical heart disease, that is characterized by (a) a short QT interval (Fig. 33.1) and (b) paroxysmal atrial and/or ventricular tachyarrhythmias resulting from an accelerated cardiac (atrial and ventricular) repolarization due to congenital (genetically heterogeneous) cardiac channelopathies. It requires exclusion of patients with secondary short QT interval [19], and without documentation of associated arrhythmogenic complications in the patient or the patient’s family a short QT interval is only an ECG abnormality. Unexplained sudden cardiac death (SCD) in a family with members having SQTS would normally be considered a manifestation of SQTS unless proven otherwise.
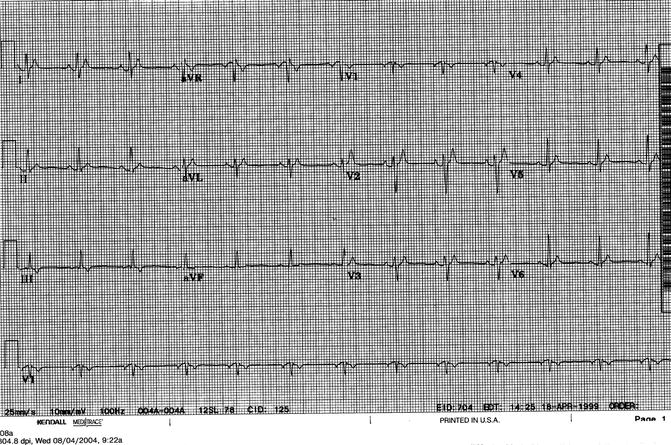
Figure 33–1.
Twelve-lead ECG from a patient with SQTS based upon a short QT interval and paroxysmal atrial fibrillation. The ECG shows sinus rhythm at a heart rate of 75 beats/min. The QT interval is 240 ms
Before the discovery of SQTS there were no well-defined lower limit for the QT interval, and normal limit for the QT interval was usually given only as an upper limit. Based upon data from Rautaharju et al. study from 1992 of the QT interval in 14,379 healthy individuals, the lower limit for the QT interval (two standard deviations below the mean predicted value) at a heart rate of 60 bpm was 360 ms [20]. More recent studies of apparently normal people have found variable results for the lower limit of QTc probably influences by the population studied and the method used for measuring the interval [11, 14, 21–25], but arguments could be made for a lower limit of the QTc interval of ≤360 ms for men and ≤370 ms for women realizing that these are approximate figures and not a great help in finding patients with SQTS. In our review of the worldwide population of patients with SQTS in 2008 [26] we found among 49 published cases the mean QT interval to be 282 ± 63 (range: 210–340) ms and the mean QTc to be 305 ± 42 (range: 248–345) ms. In a 2011 follow-up study from Europe [18] of 49 patients the QTc was 314 ± 46 ms. In recent years some patients with longer QT intervals have been published as having SQTS (vide infra). Since the QT interval in patients with SQTS varies very little with changes in heart rate [7, 27, 28], it is important to realize, that normal correction formulas for the QT interval at heart rates greater than 60 bpm will grossly overestimate, what the OT interval will be at a heart rate of 60 bpm. Paradoxically, in patients with SQTS the QTc varies with HR. The faster the HR the longer the QTc and at heart rates 80–90 bpm many patients with SQTS will have a normal QTc. At heart rates much above 60 bpm, as often seen in children, there is, therefore, a risk of missing patients with SQTS, when QT correction formulas are used. Therefore, SQTS cannot be defined by QTc. In order to get a reliable assessment of the QT interval in a patient suspected of having a short QT interval and possibly SQTS every effort should be made to get an ECG at a heart rate as close to 60 bpm as possible, even by using a drug if necessary. The lack of adaptation of QT interval to change in HR in patients with SQTS has been suggested as a diagnostic tool in work up of patients suspected of having SQTS. It is likely, however, that anybody with a short QT interval will have a QT/RR slope lower than people with normal QT intervals. More research in this area is needed before it can be recommended as a reliable diagnostic tool especially in borderline cases, where it would be most needed. Another common finding in the ECG of patients with SQTS in particularly in precordial leads, is tall, peaked T waves, most commonly symmetrical, but in patients with KCNJ2 mutation asymmetrical with a steep descent [29, 30]. Some studies have pointed out, that the Tpeak−Tend interval and Tpeak−Tend/QT ratio is prolonged in patients with SQTS compatible with an augmented transmural dispersion of repolarization [6, 18].
Since the discovery of SQTS and the significance of a short QT interval the question has been: “when does a short QT interval become clinically significant?”
The first indication of the prognostic significance of a relatively short QT interval was from a study in 1993 by Algra et al. [31]. Out of 6,693 patients who underwent 24 h Holter monitoring and followed for 2 years, patients with a QTc < 400 ms had a 2.4-fold increase in sudden death rate compared to patients with a QTc of 400–440 ms. This was slightly more than patients with a QTc > 440 ms, who had a 2.3-fold increase. The authors argued for the possibility of their finding being a true pathophysiological phenomenon with a relative short QT interval possibly leading to life-threatening arrhythmias.
Evidence that shortening of the QT interval may play a role for the occurrence of idiopathic VT was provided by Fei and Camm in 1995 [32]. Twenty-four hour Holter monitoring was used to detect 60 episodes of monomorphic repetitive ventricular tachycardia in ten patients. Analysis of three consecutive QT intervals immediately before the onset of ventricular tachycardia found these QT intervals significantly shorter than the intervals measured 40 min before at the same heart rates (342 ± 34 vs. 353 ± 35 ms, P < 0.001). Of the 60 episodes the QT intervals were shortened in 45 (75 %) compared to the intervals 40 min earlier. The shortening was explained by sudden parasympathetic withdrawal leading to sympathetic predominance and thereby QT shortening. The shortening was suggested by the authors to play an important role in the pathogenesis of idiopathic VF.
In 1999 we presented a case of paradoxical shortening of the QT interval to 216 ms during severe transient bradycardia in a child with recurrent cardiac arrest and discussed deceleration-dependent shortening of the QT interval as a trigger of arrhythmic events [33]. We proposed activation of Ik Ach due to an unusually high vagal discharge to the heart as a possible mechanism responsible for both slowing of the heart rate and shortening of the QT interval.
Visken et al. [34] compared ECGs of 28 patients with idiopathic VF (17 men and 11 women, age 31 ± 17 years) to those of 270 age- and gender- matched healthy controls. They found that the QTc of males with idiopathic VF was shorter than the QTc of healthy males (371 ± 22 ms vs. 385 ± 19 ms, P = 0.034), and 35 % of the male patients had QTc < 360 ms (range 326–350 ms) compared to only 10 % of male controls (345–360 ms). However, no such differences were found among women. They suggested that QTc intervals shorter than 360 ms might entail some arrhythmic risk.
The limited data available seem to indicate, that the extent of QT interval shortening is associated with the probability of an adverse outcome. In the largest study so far by Giustetto et al. [18] the QTc interval was 300 ± 20 ms in ten patients with cardiac arrest and 309 ± 19 ms in 19 patients with no such event. In our review of the world-wide population of patients with SQTS [26], in 16 patients with SCD or aborted SCD the QT intervals were 271 ± 33 ms compared to 291 ± 55 ms in 26 patients without SCD, aborted SCD and atrial fibrillation. In seven patients with atrial fibrillation only, the QT intervals were 265 ± 49 ms. From these data it is also apparent that the great majority of patients published so far with SQTS have had very short QT/QTc intervals, but because of sporadic cases with somewhat longer QT intervals [12, 28] we are not at a point where the diagnosis can be based upon the duration of the QT interval alone. Even though recently published proposed diagnostic criteria for the diagnosis of SQTS [17] was met by some criticism [35, 36], they very well points out features besides the QT interval, that has to be taken into consideration when making a diagnosis of SQTS, such as clinical and family history in addition to genotyping if possible.
Genetic Basis of SQTS
SQTS is a genetically heterogeneous disease. In the study by Giustetto et al. [18] the yield of genetic screening in SQTS was 23 % of the investigated index patients in their study. So far a genetic mutation has been found in at least 22 patients from 13 families with SQTS [4, 9, 13, 29, 37–40], and in 15 patients from five families with a short QT interval and ST-segment changes (Table 33.2) [12, 41, 42]. Patients in the later group have ST changes in precordial leads somewhat similar to Brugada Syndrome patients either spontaneously or by provocation, and since there is no clear basis on which to select one syndrome over another, they have been looked upon as a distinct clinical entity. With QTcs ranging from 330 to 360 ms the QT interval in these patients has generally not been as short as reported for patients with only SQTS. Initially mutations in patients with SQTS were found in three genes (KCNH2, KCNQ1 and KCNJ2) encoding for potassium channels and the respective syndromes were called SQT1, SQT2 and SQT3 based upon the chronology of their discovery.
Table 33–2.
Gene mutations associated with SQTS
Mutation | Gene | # Families | # Patients |
|---|---|---|---|
Gene mutations associated with SQTS | |||
N588K | KCNH2 | 5 | ≥12 |
T618I | KCNH2 | 2 | ≥2 |
E50D | KCNH2 | 1 | 2 |
V307L | KCNQ1 | 1 | 1 |
V141M | KCNQ1 | 2 | 2 |
I274V | KCNQ1 | 1 | 1 (SIDS) |
D172N | KCNJ2 | 1 | 2 |
Gene mutations associated with Short QT and ST-segment changes | |||
R1135H | KCNH2 | 1 | 3 |
S481L | CACNB2 | 1 | 6 |
G490R | CACNA1C | 1 | 2 |
A39V | CACNA1C | 1 | 1 |
S755T | CACNA2D1 | 1 | 3 |
In the first two families from Europe with SQT1 reported in 2004 by Gaita et al. [2] two different missense mutations (C1764A and C1764G) were later discovered and found to result in the same amino acid change (N588K) in the S5-P loop region of the cardiac I Kr channel KCNH2 (HERG) [4]. Within the same year the first patient with SQT2 was reported, when a mutation (V307L) in the KCNQ1 gene encoding the I Ks channel KvLQT1 was found in a 70 year old male with idiopathic ventricular fibrillation and a short QT interval [38]. Another KCNQ1 mutation was later found in two unrelated patients with bradycardia in utero and born with atrial fibrillation and high degree atrio-ventricular block in addition to a very short QT interval [9]. Genetic testing showed a missense mutation, G to A substitution at nucleotide 421 (g421a). This mutation results in substitution of valine by methionine at position 141 (V141M) adjacent to a previously described S140G mutation for familial AF [43]. Finally, in 2007 a third gain-of-function mutation (I274V) was found in KCNQ1 in a patient with SIDS [44].
In 2005 in an Italian family a KCNJ2 gene mutation was found in a 5-year old girl with a short QT interval (SQT3). Genetic analysis by Priori et al. led to the identification of a single base pair substitution (G514A) in KCNJ2, resulting in an amino acid change from aspartic acid to asparagine at position 172 in the Kir2.1 potassium channel (IK1) [29]. From these initial findings an interesting concept emerged, that LQTS and SQTS had closely related genetic basis and could be considered “allelic diseases”. Indeed, all three SQTS genes were known to also cause LQTS. Since then eight additional mutations responsible for shortening of the QT interval have been found (Table 33.2).
Cellular Basis of Arrhythmogenesis in SQTS
Mutations effecting potassium channels all leads to a gain-of-function. There is general agreement that gain-of-function from the N588K-HERG mutation in patients with SQT1 leading to an increase in the repolarizing currents active during phase 2 and 3 of the AP stems from severely compromised inactivation. McPate et al. found a ∼ +60 mV positive-shift in voltage dependence of IHERG inactivation [45]. Not all cells are affected to the same degree and Cordero et al. found that ventricular Purkinje cells were minimally affected [46]. Grunnet et al.’s patch clamp experiments showed that the biophysical characterization of the short QT mutation HERG-N588K was compatible with a mixed gain-and loss-of-function [47]. The most prominent loss-of-function property was reduced tail currents, but also slower activation and faster deactivation kinetics leading to reduced ability to conduct current at the end of repolarization. The authors pointed out that in patients carrying HERG-N588K the loss-of-function of repolarization current and diastolic HERG current might be at least as pro-arrhythmic as the gain-of-function of plateau current. All information on the likely consequences for I Kr -kinetics of N588K-HERG mutation comes from studies of the HERG1a isoform. Recent evidence suggests, however, that native cardiac I Kr may not be comprised of HERG1a alone, but rather of HERG1a heteromerically expressed with an alternative transcript, HERG1b, an isoform with a truncated N-terminus. This lead McPate el al. to conduct a study to determine the effects of the N588K-HERG SQT1 mutation on co-expressed HERG1a/1b channels [48]. Their data showed that the inactivation-attenuation effects of the N588K mutation were markedly greater when co-expressed HERG1a and 1b were studied, than when HERG1a alone was studied. The study also confirmed the differential effect the N588K-HERG mutation has on current during ventricular and Purkinje AP’s as initially suggested by data from the study by Cordeiro et al. [46] and McPate et al. [45].
Another mutation, which effects the plateau phase of the action potential is V307L-KCNQ1 seen in SQT2 patients. Bellocq et al. did conventional patch-clamp experiments using COS-7 cells and showed faster activation at more negative potentials for V307L channels compared to wild-type (WT) while kinetics for deactivation were similar [38]. Computer simulations with findings from the patch-clamp experiments incorporated clearly showed diminished AP duration favoring the association of a short QT interval with the V307L-KCNQ1 mutation. Harchi et al. using perforated-patch voltage-clamp recordings (Chinese Hamster Ovary cells) at 37 °C of whole-cell current with epicardial ventricular AP waveform carried by co-expressed KCNQ1 and KCNE1 showed a marked (−36 mV) shift in half-maximal activation for V307L compared to WT-KCNQ1 [49]. In contrast to Bellocq et al. [38], they also found a significant slowing of current deactivation. They also looked at the effect of the mutation on atrial cells and found peak repolarising current significantly augmented for V307L-KCNQ1 compared to WT for both ventricular and atrial AP commands, consistent with an ability of the V307L mutation to increase repolarising I Ks in both regions. Although atrial fibrillation was not reported for the patient in whom the V307L-KCNQ1 mutation was first identified [38], SQTS patients especially with the N508K-HERG mutation do experience atrial fibrillation at a higher incidence than expected by chance alone [18]. Atrial fibrillation was also part of the clinical picture in patients with the V141M-KCNQ1 mutation [9, 26]. These patients presented with bradycardia in utero and were born with a short QT interval and atrial fibrillation with a very slow heart rate suggesting high degree AV block. The inability to restore sinus rhythm in one of the patient and the inability to maintain sinus rhythm for more than a few hours in the other patient suggested possibly sick sinus syndrome as well. Hong et al. [9] injected oocytes from Xenopus laevis with WT or V141M–KCNQ1 cRNA with or without KCNE1 cRNA and 2–3 days later exposed them to two electrode voltage clamp recordings. The V141M mutation did not noticeably alter the gating of KCNQ1 channels expressed alone in oocytes. The WT-KCNQ1/KCNE1 channels exhibited a voltage-dependent threshold of activation near −50 mV and activated very slowly. In sharp contrast, the V141M–KCNQ1/KCNE1 channel current developed instantly at all voltages tested, consistent with the interpretation that these channels were constitutively open. Computer modeling showed decrease in peak voltage and shortening of APD consistent with shortening of the QT interval. The effect of the V141M gain of function mutation was also modeled in a computer model of rabbit sinoatrial node cells. The results indicated that the enhanced outward I Ks causes cessation of spontaneous activity and a stabilization of the resting membrane potential at a level positive to the normal maximum diastolic potential of these cells. The exact mechanism behind the bi-nodal dysfunction seen in these patients needs, however, further exploration. A third mutation in KCNQ1 has been considered a possible cause for sudden infant death syndrome. In a patient with sudden infant death Arnestad et al. [13] found a 1,274 V-KCNQ1 mutation and Rhoades et al. [44] presented electrophysiological data from patch clamp recordings (Chinese hamster ovary cells) showing that I274V-KCNQ1 in the presence of KCNE1 causes gain of function in I Ks characterized by increased current density, faster activation, slower deactivation, and accumulation of instantaneous current during repeated stimulation. To test the hypothesis that I274V may promote a short QT syndrome phenotype, computerized modelings of ventricular action potentials were performed comparing WT-Iks to heterozygous I274V-Iks. At all cycle length the AP was shorter for I274V-Iks supporting the prediction that I274V-KCNQ1 will cause a short QT phenotype, and, therefore, may be a plausible explanation for sudden death in an infant carrying this mutation.
SQT3 patients are characterized by a mutation in the KCNJ2 gene [29]. Whole-cell patch-clamp studies of the heterologously expressed human D172N channel have demonstrated larger outward IK1 than the WT at potentials between −75 and −45 mV, with the peak current being shifted in the former with respect to the later (WT, −75 mV; D172N and −65 mV). Co-expression of WT and mutant channels to mimic heterozygous condition of the proband has yielded an outward current that is intermediate between WT and D172N. The authors hypothesized that the tall and asymmetrical T-waves with an exceedingly rapid terminal phase seen in these patients and not seen in SQT1 or SQT2 patients might be related to a more sudden acceleration of the final phase of action potential repolarization in patients with the D172N mutation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


